




Abstract
PHAST (PHAge Search Tool) and its successor PHASTER (PHAge Search Tool – Enhanced Release) have become two of the most widely used web servers for identifying putative prophages in bacterial genomes. Here we review the main capabilities of these web resources, provide some practical guidance regarding their use and discuss possible future improvements. PHAST, which was first described in 2011, made its debut just as whole bacterial genome sequencing and was becoming inexpensive and relatively routine. PHAST quickly gained popularity among bacterial genome researchers because of its web accessibility, its ease of use along with its enhanced accuracy and rapid processing times. PHASTER, which appeared in 2016, provided a number of much-needed enhancements to the PHAST server, including greater processing speed (to cope with very large submission volumes), increased database sizes, a more modern user interface, improved graphical displays and support for metagenomic submissions. Continuing developments in the field, along with increased interest in automated phage and prophage finding, have already led to several improvements to the PHASTER server and will soon lead to the development of a successor to PHASTER (to be called PHASTEST).
Introduction
Bacteriophages are the most abundant biological entities on Earth. They are recognized as a major contributor to microbial genetic variation and diversity [1]. Bacteriophages are known to play important roles in marine carbon and nutrient cycling [2], and can provide bacteria with the ability to become pathogenic, resist antibiotics or adapt to new ecological niches [1, 3]. A key part of the bacteriophage life cycle, lysogeny, involves integrating the phage genome into the host bacterial chromosome at well-defined insertion points. These latent phages are called prophages and in some cases prophages can become permanently embedded into the bacterial genome, becoming cryptic prophages [4]. These cryptic prophages often serve as genetic ‘fodder’ for future evolutionary changes of the host bacterium [5]. Prophages and cryptic prophages can account for up to 20% of the genetic material in some bacterial genomes [3]. With the ever-increasing number of newly sequenced bacterial genomes and the recognition that prophage sequences account for a significant portion of bacterial DNA, software for performing prophage and cryptic prophage identification in bacterial genomes has become essential to many bacterial genome annotation pipelines.
However, detecting prophages in the complex milieu of bacterial DNA is a challenging computational problem. Early approaches to identify prophages included those based on atypical nucleotide content or the identification of disrupted genes [6–7], but unfortunately these simple approaches proved to be too unreliable [8]. In the late 2000s a number of improved programs and web servers were introduced to help find prophage sequences within bacterial genomes. These included Phage_Finder [9], Prophage Finder [10], and Prophinder [11], which employed sequence matching to known phage and bacterial genes, tRNA and dinucleotide analysis, along with attachment site detection using hidden Markov models. These programs greatly improved prophage prediction accuracy, and inspired the development of even more prophage finding tools including ones that avoid reliance on known phage sequences [12] or designed specifically for metagenomic sequencing data [13] (see Table 1 for more information). Even with these advances, there was still room for improvement, chiefly in the areas of speed and usability. It is precisely these areas that motivated our development of two new tools for prophage annotation: PHAST (PHAge Search Tool), published in 2011 [16] and its successor PHASTER (PHAge Search Tool – Enhanced Release), released in 2016 [18].
A summary of different prophage finding tools and methods
| Program/method (year published) [Reference] | Input data requirements | Matching to known phage sequences | Prophage detection method | Platform |
|---|---|---|---|---|
| Dinucleotide abundance (2002) [6, 7] | Unannotated FASTA | None | Dinucleotide relative abundance | NA |
| Phage_Finder (2006) [9] | Pre-annotated contigs accepted | HMMER [14], BLASTP [15] | Knowledge-based rules/metrics, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| Prophage Finder (2006) [10] | Unannotated FASTA | BLASTX [15] | Statistical metrics | Web-based (no longer active) |
| Prophinder (2008) [11] | Pre-annotated | BLASTP | Statistical metrics | Web service via Perl script run on Unix-based OSs |
| PHAST (2011) [16] | Unannotated FASTA or pre-annotated | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| PhiSpy (2012) [12] | Pre-annotated | None | Similarity-agnostic statistical metrics, phage insertion point, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| VirSorter (2015) [13] | Unannotated FASTA contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | CyVerse discovery environment [17] |
| PHASTER (2016) [18] | Unannotated FASTA or pre-annotated contigs accepted | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| VRprofile (2017) [19] | Unannotated FASTA or pre-annotated contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | Web-based |
| Program/method (year published) [Reference] | Input data requirements | Matching to known phage sequences | Prophage detection method | Platform |
|---|---|---|---|---|
| Dinucleotide abundance (2002) [6, 7] | Unannotated FASTA | None | Dinucleotide relative abundance | NA |
| Phage_Finder (2006) [9] | Pre-annotated contigs accepted | HMMER [14], BLASTP [15] | Knowledge-based rules/metrics, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| Prophage Finder (2006) [10] | Unannotated FASTA | BLASTX [15] | Statistical metrics | Web-based (no longer active) |
| Prophinder (2008) [11] | Pre-annotated | BLASTP | Statistical metrics | Web service via Perl script run on Unix-based OSs |
| PHAST (2011) [16] | Unannotated FASTA or pre-annotated | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| PhiSpy (2012) [12] | Pre-annotated | None | Similarity-agnostic statistical metrics, phage insertion point, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| VirSorter (2015) [13] | Unannotated FASTA contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | CyVerse discovery environment [17] |
| PHASTER (2016) [18] | Unannotated FASTA or pre-annotated contigs accepted | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| VRprofile (2017) [19] | Unannotated FASTA or pre-annotated contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | Web-based |
A summary of different prophage finding tools and methods
| Program/method (year published) [Reference] | Input data requirements | Matching to known phage sequences | Prophage detection method | Platform |
|---|---|---|---|---|
| Dinucleotide abundance (2002) [6, 7] | Unannotated FASTA | None | Dinucleotide relative abundance | NA |
| Phage_Finder (2006) [9] | Pre-annotated contigs accepted | HMMER [14], BLASTP [15] | Knowledge-based rules/metrics, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| Prophage Finder (2006) [10] | Unannotated FASTA | BLASTX [15] | Statistical metrics | Web-based (no longer active) |
| Prophinder (2008) [11] | Pre-annotated | BLASTP | Statistical metrics | Web service via Perl script run on Unix-based OSs |
| PHAST (2011) [16] | Unannotated FASTA or pre-annotated | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| PhiSpy (2012) [12] | Pre-annotated | None | Similarity-agnostic statistical metrics, phage insertion point, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| VirSorter (2015) [13] | Unannotated FASTA contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | CyVerse discovery environment [17] |
| PHASTER (2016) [18] | Unannotated FASTA or pre-annotated contigs accepted | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| VRprofile (2017) [19] | Unannotated FASTA or pre-annotated contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | Web-based |
| Program/method (year published) [Reference] | Input data requirements | Matching to known phage sequences | Prophage detection method | Platform |
|---|---|---|---|---|
| Dinucleotide abundance (2002) [6, 7] | Unannotated FASTA | None | Dinucleotide relative abundance | NA |
| Phage_Finder (2006) [9] | Pre-annotated contigs accepted | HMMER [14], BLASTP [15] | Knowledge-based rules/metrics, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| Prophage Finder (2006) [10] | Unannotated FASTA | BLASTX [15] | Statistical metrics | Web-based (no longer active) |
| Prophinder (2008) [11] | Pre-annotated | BLASTP | Statistical metrics | Web service via Perl script run on Unix-based OSs |
| PHAST (2011) [16] | Unannotated FASTA or pre-annotated | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| PhiSpy (2012) [12] | Pre-annotated | None | Similarity-agnostic statistical metrics, phage insertion point, gene function | Unix-based OSs (e.g. Linux, MacOS) (download) |
| VirSorter (2015) [13] | Unannotated FASTA contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | CyVerse discovery environment [17] |
| PHASTER (2016) [18] | Unannotated FASTA or pre-annotated contigs accepted | BLASTP | Knowledge-based rules/metrics, gene function | Web-based |
| VRprofile (2017) [19] | Unannotated FASTA or pre-annotated contigs accepted | HMMER, BLASTP | Statistical metrics, gene function | Web-based |
In this article, we review some of the key features that have made PHAST and PHASTER popular and effective tools for bacterial prophage identification and prediction. We also provide some practical guidance regarding their use and summarize some of the more recent improvements to the PHAST and PHASTER servers. Additionally, future directions for improving their capabilities are discussed with the aim of releasing the ‘PHASTEST’ and most accurate prophage prediction server in the near future.
Phast
PHAST was released in 2011 [16], at a time when next-generation sequencing was making bacterial genome sequencing fast, inexpensive and increasingly routine. Partly because of its good timing and partly because of its speed and ease of use, PHAST quickly became one of the most popular tools for finding prophage sequences in bacterial genomes. In addition to being an easy-to-use web server, PHAST also offered something relatively unique to prophage servers. That is, it was capable of automatically annotating raw bacterial DNA sequence data. Apart from Prophage Finder [10], earlier prophage finding programs had required that input genomes be pre-annotated with open reading frames (ORFs) and/or have their tRNA sites already identified. This required users to perform additional time-consuming operations before running their sequences through a phage finding program. PHAST eliminated that extra step. While PHAST still accepts pre-annotated genomes in GenBank format, users only need to submit the raw DNA sequence of a bacterial genome in FASTA format, as PHAST will perform its own gene prediction and tRNA/tmRNA annotation. Over the past 5 years, roughly 85% of uploaded submissions to PHAST (and PHASTER) have been raw genomic DNA sequences.
PHAST also offered a number of other features than made it particularly easy and convenient to use. For instance, it produced detailed, web-browsable prophage annotations that allowed users to examine their results using a graphical genome viewer. Additionally, PHAST provided both downloadable graphics and a downloadable text-based version of its prophage predictions (see Figure 1 for a montage of PHAST output images). PHAST also supported batch submissions of up to 10 genomes at a time through its web interface, as well as an API (Application Programming Interface) through which users could submit larger batch submissions of hundreds or even a few thousand sequences. After PHAST’s publication, a database of PHAST predictions for thousands of public bacterial genomes was developed and made available as well. These pre-calculated predictions could be retrieved by entering a genome’s GenBank accession number.

A montage of PHAST output images. A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
Last but not least, PHAST was much faster than previous leading methods. More specifically, PHAST was able to find prophages in a typical bacterial genome in about 3 min whereas most other programs could take 30 min to 2 h. At the same time, PHAST was found to be more accurate than competing methods. When benchmarked against a set of 54 manually curated bacterial genomes [3], PHAST achieved sensitivity and positive predictive value (PPV) measures of 79.4% and 86.5%, respectively, on raw sequence input, and 85.4% sensitivity and 94.2% PPV on pre-annotated genomes from GenBank [16]. On the other hand, other programs either had sensitivities 8% lower or worse (with comparable PPVs), or achieved higher sensitivity only at the cost of much higher false positive rates [16]. PHAST also predicted the completeness of predicted prophages, scoring them as: (i) intact, (ii) incomplete or (iii) questionable. The combination of speed, accuracy and usability has made PHAST a very appealing tool for finding prophages in bacterial genomes.
The prophage prediction pipeline used by PHAST can be outlined as follows. Raw genomic sequence input is first annotated using the GLIMMER gene prediction software [20], and tRNA and tmRNA sites are found using tRNAscan-SE [21] and ARAGORN [22]. If a pre-annotated GenBank file is used, these steps are skipped. Predicted genes are searched against PHAST’s database of prophage protein sequences using BLAST, and matched genes within a minimum distance of each other are then iteratively clustered together using the DBSCAN algorithm [23]. Candidate prophage regions are then created by grouping together multiple viral gene clusters containing at least four viral genes. This process considers all possible group combinations involving one or more clusters (up to a maximum length). Candidate prophage regions are scored based on: (i) the cumulative length of the gaps between component clusters; (ii) the number and proportion of genes matching known prophage genes; (iii) the presence of BLAST hits (among the multiple BLAST hits for each gene across a candidate prophage region) that match a high proportion of a particular phage strain’s known genes and (iv) the presence of insertion sequences and matching viral genes with key functions including integrases, transposases and structural genes. Candidate prophages that score higher than alternative candidates and above a minimum threshold are chosen as PHAST’s prophage predictions. A scan is then performed for possible attachment sites, and the best candidates are predicted. Genes within predicted prophage regions that did not match known viral genes are searched against a bacterial protein sequence database in order to complete the prophage annotation.
Phaster
Although PHAST was one of the fastest and most accurate phage finding tool at the time of its publication in 2011, increasing sequence database sizes, increased user volume and numerous requests for additional features led to a significant PHAST update in 2016. Therefore, an enhanced version of PHAST was created and released in 2016 [18]. This newer, better version was named PHASTER (PHAge Search Tool—Enhanced Release). While the original PHAST server is still maintained (to accommodate legacy requests and re-runs), we strongly encourage current PHAST users to migrate to the PHASTER server. Some of the reasons for moving to the PHASTER server are outlined below.
The massive increase in the number of sequenced bacterial genomes, as well as the number of known phages and prophages since PHAST’s original release have led to a significant increase (∼4×) in processing times in the PHAST pipeline. Increased interest in phage/prophage research also appears to have contributed to the high traffic levels seen by the PHAST server. To accommodate the increased computational demands, the computing cluster used by PHASTER for gene prediction and BLAST searches has been expanded 3.5-fold from 32 cores (in PHAST) to 112 CPU cores (in PHASTER). Algorithmic efficiency changes were also implemented to improve PHASTER’s performance and enable faster processing of heavy submission loads. These changes included more intelligent distribution of BLAST searches across computing nodes and a redundancy reduction in PHASTER’s bacterial protein sequence database using CD-HIT [24]. With these and other efficiency improvements, the time taken by the pipeline’s BLAST searches—which represent the lion’s share of its processing time—were reduced almost by half, without having to switch to using a faster but less accurate sequence alignment tool. In all, PHASTER’s improvements made it 4–5× faster on raw sequence submissions, without any compromise in accuracy. Indeed, as noted previously, parameter adjustment and expanded databases led to a notable increase in PHASTER’s sensitivity on raw genome submissions (from 79.4% to 85.0%) without any increase in the rate of false positives (i.e. the PPV improved from 86.5% to 87.3%) [18].
In addition to the cluster and algorithmic updates, a faster, dedicated front-end server was implemented as a Ruby on Rails application with a well-managed queuing system using Sidekiq. This allowed the PHASTER user interface to become more modern, more user friendly and more visually appealing (see Figure 2). A new detailed genome viewer compatible with modern web browsers was also implemented to allow users to examine phage details on an interactive circular genome layout. A linear view also provides additional protein information. Unlike PHAST, PHASTER allows users to save their submissions via a cookie-based system or to bookmark their results for later viewing. In addition, PHASTER stores prophage prediction results for more than 14 000 bacterial genomes, and makes them available in a fast-loading and searchable view.
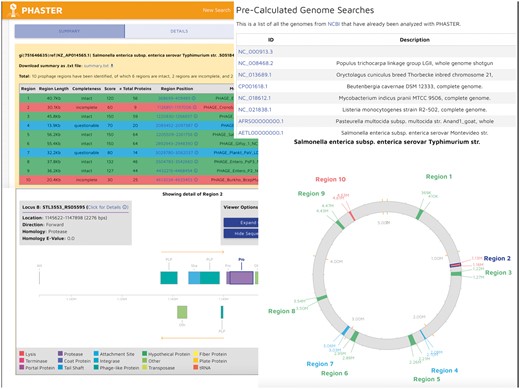
A screenshot of PHASTER’s modernized user interface. A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
In addition to faster and more accurate prophage predictions, PHASTER also has an important new feature for processing assembled contigs from metagenomic input. Metagenomic data with pre-assembled contigs is handled efficiently by the PHASTER pipeline, which is able to find prophage regions within individual contigs of at least 2000 bp. Genes are predicted using FragGeneScan [25], designed for finding genes in fragmentary metagenomic sequences, and the updated genome viewer displays individual contigs containing predicted prophage regions. The PHASTER API also supports the submission of metagenomic contigs without using the web interface. Importantly, the original PHAST API has been re-designed for PHASTER to allow users to upload a large number of submissions to the server and check the status of each job at their convenience, be they genomic sequences or metagenomic contigs. As with PHAST, results from PHASTER queries can be downloaded via the API or viewed on the interactive web interface. Table 2 compares PHAST with PHASTER.
A feature comparison between PHAST and PHASTER
| Feature | PHAST (as of January 2011) | PHASTER (as of May 2017) |
|---|---|---|
| Viral sequence database | ∼45 000 sequences | ∼230 000 sequences |
| Bacterial sequence database | ∼4 million sequences | ∼9 million sequences, streamlined through CD-HIT filtering |
| Computing cluster | 32 CPU cores | 192 CPU cores |
| BLAST | Legacy version 2.2.16 | BLAST+ version 2.3.0+ |
| Cluster use optimization | Rudimentary | Smart partitioning of query sequences and target bacterial DB; optimized execution parameters |
| Front-end server | Shared, single CPU | 50% faster, dedicated |
| Front-end website | Perl and CGI | Ruby on Rails |
| Genome viewer | Adobe Flash | JavaScript, AngularPlasmid and D3 |
| Queuing system | Flat file | Uses Sidekiq for threading submissions |
| Recall previous user submissions | Bookmark page | ‘My Searches’ feature or bookmark |
| Pre-computed genome results for quick query searching | 0 | >14 000 |
| Retrieve previously annotated genome results | GenBank accession or GI number only | GenBank accession, GI number, or full sequence |
| Metagenomic data handling | NA | Supported |
| Feature | PHAST (as of January 2011) | PHASTER (as of May 2017) |
|---|---|---|
| Viral sequence database | ∼45 000 sequences | ∼230 000 sequences |
| Bacterial sequence database | ∼4 million sequences | ∼9 million sequences, streamlined through CD-HIT filtering |
| Computing cluster | 32 CPU cores | 192 CPU cores |
| BLAST | Legacy version 2.2.16 | BLAST+ version 2.3.0+ |
| Cluster use optimization | Rudimentary | Smart partitioning of query sequences and target bacterial DB; optimized execution parameters |
| Front-end server | Shared, single CPU | 50% faster, dedicated |
| Front-end website | Perl and CGI | Ruby on Rails |
| Genome viewer | Adobe Flash | JavaScript, AngularPlasmid and D3 |
| Queuing system | Flat file | Uses Sidekiq for threading submissions |
| Recall previous user submissions | Bookmark page | ‘My Searches’ feature or bookmark |
| Pre-computed genome results for quick query searching | 0 | >14 000 |
| Retrieve previously annotated genome results | GenBank accession or GI number only | GenBank accession, GI number, or full sequence |
| Metagenomic data handling | NA | Supported |
A feature comparison between PHAST and PHASTER
| Feature | PHAST (as of January 2011) | PHASTER (as of May 2017) |
|---|---|---|
| Viral sequence database | ∼45 000 sequences | ∼230 000 sequences |
| Bacterial sequence database | ∼4 million sequences | ∼9 million sequences, streamlined through CD-HIT filtering |
| Computing cluster | 32 CPU cores | 192 CPU cores |
| BLAST | Legacy version 2.2.16 | BLAST+ version 2.3.0+ |
| Cluster use optimization | Rudimentary | Smart partitioning of query sequences and target bacterial DB; optimized execution parameters |
| Front-end server | Shared, single CPU | 50% faster, dedicated |
| Front-end website | Perl and CGI | Ruby on Rails |
| Genome viewer | Adobe Flash | JavaScript, AngularPlasmid and D3 |
| Queuing system | Flat file | Uses Sidekiq for threading submissions |
| Recall previous user submissions | Bookmark page | ‘My Searches’ feature or bookmark |
| Pre-computed genome results for quick query searching | 0 | >14 000 |
| Retrieve previously annotated genome results | GenBank accession or GI number only | GenBank accession, GI number, or full sequence |
| Metagenomic data handling | NA | Supported |
| Feature | PHAST (as of January 2011) | PHASTER (as of May 2017) |
|---|---|---|
| Viral sequence database | ∼45 000 sequences | ∼230 000 sequences |
| Bacterial sequence database | ∼4 million sequences | ∼9 million sequences, streamlined through CD-HIT filtering |
| Computing cluster | 32 CPU cores | 192 CPU cores |
| BLAST | Legacy version 2.2.16 | BLAST+ version 2.3.0+ |
| Cluster use optimization | Rudimentary | Smart partitioning of query sequences and target bacterial DB; optimized execution parameters |
| Front-end server | Shared, single CPU | 50% faster, dedicated |
| Front-end website | Perl and CGI | Ruby on Rails |
| Genome viewer | Adobe Flash | JavaScript, AngularPlasmid and D3 |
| Queuing system | Flat file | Uses Sidekiq for threading submissions |
| Recall previous user submissions | Bookmark page | ‘My Searches’ feature or bookmark |
| Pre-computed genome results for quick query searching | 0 | >14 000 |
| Retrieve previously annotated genome results | GenBank accession or GI number only | GenBank accession, GI number, or full sequence |
| Metagenomic data handling | NA | Supported |
Using the PHAST and PHASTER servers—practical considerations
In this section, we discuss a few tips and practical considerations for using the PHAST and PHASTER websites. PHAST and PHASTER can be accessed online at http://phast.wishartlab.com and http://phaster.ca, respectively. A video tutorial/overview of PHASTER is also now available on the PHASTER homepage.
Input choices matter
Both PHAST and PHASTER can accept raw genomic DNA sequences in FASTA format, as well as pre-annotated genomic sequences in GenBank format. As noted above, using the pre-annotated genomes from GenBank generally improves the performance of PHAST and PHASTER. Our latest data shows the sensitivity and PPV improve by 1.9% and 3.7%, respectively, when the pre-annotated genomes from GenBank are used in PHASTER [18]. Of course, many PHAST/PHASTER users are working with newly sequenced organisms and so they are not able to get pre-annotated GenBank files. Nevertheless, if users wish to get the very best results, we recommend that they try to obtain (via GenBank) or try to perform the most thorough genome annotation possible prior to running their data through PHAST or PHASTER. Good quality input leads to better quality output.
For multiple sequence submission, PHAST and PHASTER allow users to submit up to 10 sequences at a time via the web interface in batch mode. In PHASTER, users may also submit contigs assembled from metagenomic sequencing data. There is no limit on the number of contigs, but only those >2000 bp will be processed. Batch genome submissions are distinguished from contig submissions automatically based on analysis of sequence length distributions.
Understanding prophage prediction results
While both PHAST and PHASTER are excellent tools for predicting the presence of prophage regions, it is important to remember that there is always going to be some uncertainty with regard to the exact extent of a predicted prophage. This includes the location of predicted attachment sites. For example, sometimes potential phage genes are found beyond the bounds marked by potential attachment sites, and it is not clear whether the phage genes or attachment sites are erroneous, or whether there may be a series of prophage regions, or whether an ‘intact’ prophage may be nested within an ancient degraded prophage resulting in a ‘stacked’ region. PHAST and PHASTER do not try to decide which is the case, and therefore they will predict attachment sites at ‘odd’ locations, leaving it to the user to make a final determination.
The sensitivity of the clustering/grouping algorithms (see above) to the local density of predicted genes with matches to known phage genes can also impact the marked extent of predicted prophages. Moreover, in the case of more marginal predictions, even small changes in phage gene density can impact whether a prophage is predicted at all. As an illustrative example, the presence of genes matched to both a known integrase and a known structural gene (e.g. encoding a capsid protein) within a candidate prophage region is normally sufficient to warrant prediction of a prophage region. If, however, these two genes are present in two separate small clusters of phage-matched genes, and there is insufficient density of phage-matched genes in between the two clusters so that they are not joined, then neither cluster (on its own) may score sufficiently well leading to no prophage prediction. This should be kept in mind, for example, when comparing the presence/absence of predicted prophages in two closely related genomes.
Using the API
Both PHAST and PHASTER provide API’s designed to be easily integrated with other programs, allowing users to automatically upload multiple submissions and retrieve the results when they are complete. Instructions for the API’s are provided on the respective ‘Instructions’ and ‘Help’ pages of the PHAST and PHASTER websites. For the PHASTER API, if an accession number is provided, previously computed results are immediately retrieved (if these exist) or a new job is queued after the corresponding GenBank file is retrieved from NCBI. If a FASTA sequence file is uploaded, a new job is created after the necessary validations are performed. In both cases, the response contains a new job identifier that users may submit later to retrieve the results. The position of the new job in the queue is also returned to give users an estimate of how long the job will take to complete. If a script is used to access the API, timed GET requests may be implemented to periodically check the status of each queued job identifier. The response for this type of request includes the job status. If the job is complete, the results are returned in standard text format. A link to the web interface is also provided should a user wish to visualize the results. When posting a sequence file, it is important to note that the API allows users to specify whether the sequence file contains metagenomic contigs as input. The option ‘contigs’ may be set in the job request to be processed as such. If this option is not specified, PHASTER will process the first sequence in the uploaded file. Thus, the API supports single-sequence uploads per request, or multiple sequences for metagenomic contigs.
Other features
PHAST and PHASTER each provide several different kinds of result sections once a query has been analyzed. The ‘Summary’ section on the web interface outlines the prophage regions identified in a given sequence, along with a completeness score and additional details. This table is downloadable in text format. The ‘Details’ section gives a more comprehensive annotation of each gene and attachment site. This table is also downloadable in text format. The ‘Genome Viewer’ outlines the prophage regions on the circular bacterial chromosome corresponding to the positions in the given raw sequence. Clicking on a region of the circular genome allows users to interactively navigate to a detailed view of the relevant genes/proteins in that region. In addition to the text result files, images from the genome viewer can be downloaded for later review. In the case of PHASTER, it is important to note that users may also save a submission via the ‘Remember Me’ option, which adds a link under the ‘My Searches’ tab that can be accessed later (from the same browser on the same machine). Results pages can also be bookmarked and returned to later with no browser/machine restrictions. A list of pre-computed results for over 14 000 bacterial genomes is also available for browsing under the ‘Genomes’ tab on the main navigation bar. Using this online database, users may wish to search for a particular bacterial genome before submitting their genome.
Recent developments and future possibilities
PHAST and PHASTER have become very popular over the past few years. They have been used by scientists from over 150 countries and together the two servers receive nearly 3000 visits every month. PHAST and PHASTER process an average of 4000 and 11 000 submissions per month, respectively. For PHASTER, this is equivalent to an average of one submission every 4 min. All of this data processing is supported by an in-house computing cluster equipped with the necessary tools to perform the gene prediction and sequence search portions of their pipelines.
We continue to make improvements to PHASTER as we receive user feedback and as we monitor its user load. For instance, since PHASTER’s publication in 2016 we have already increased its computational power by adding another 80 CPUs (increasing the cluster size from 112 to 192 CPUs). While PHASTER is being adapted to handle a growing number of user submissions, we have also aided other labs that have sought to install a standalone, parallelized PHASTER pipeline on their own infrastructure. Going forward, as bacterial sequence annotation needs to grow ever larger and as highthroughput computational resources become more widely available to researchers through cloud computing and/or government-sponsored supercomputing resources, providing better support for researchers to install standalone versions of PHASTER is now becoming a greater priority. Development of a ‘containerized’ version of PHASTER is one possibility that could allow greater portability across different high-performance computing platforms.
While PHASTER has numerous capabilities, a number of potential improvements are being considered or are under development. For instance, a video tutorial for PHASTER has recently been created that takes users through each of its operations and options. This video is now available on the PHASTER homepage. Several other usability enhancements are in the works. These include providing better support for interoperable file formats, adding the ability to explore weaker phage-related signals neighboring predicted prophage regions, tools for comparing prophage content between different bacterial genomes, and identification of additional mobile genetic elements. Incorporating a broader, formal ontology of phage protein function, such as that used in the ACLAME database [26], could augment PHASTER’s prophage annotation quality and potentially improve prophage scoring, leading to more accurate predictions. We encourage members of the research community to continue to provide feedback regarding not only PHASTER’s usability but also PHASTER’s prediction accuracy. All suggestions and any experimentally determined prophage annotations are welcome, as many will eventually be incorporated into PHASTER’s databases and programming updates.
Several other potential improvements, both nearer and longer term, relate the impact of metagenomic studies on prophage finding. The past couple years has seen the development of tools designed to better detect phage sequences in fragmented metagenomic data [18, 27]. We believe that PHASTER’s handling of metagenomic sequences could potentially be improved along similar lines. For instance, better handling of shorter contigs and the incorporation of additional metrics, such as gene length and degree of strand switching, could help improve prophage predictions in fragmentary sequence data. There have also been increasing efforts to apply metagenomic sequencing to study previously uncharacterized viruses among uncultured bacterial species. This is the so-called ‘viral dark matter’ and it represents one of the most genetically diverse and poorly understood biological entities on the planet [28, 29]. Accordingly, one possible enhancement could be to provide additional options in PHASTER to choose alternate gene finding algorithms for metagenomic data submissions, not only to enable the use of methods better suited to a user’s sequencing data quality [30], but also to provide the option of using unsupervised methods for predicting genes in novel genomes [31].
Furthermore, certain types of phages exhibit lifecycle characteristics that do not involve their integration into host bacterial genomes. ‘Extrachromosomal prophages’ or ‘plasmid prophages’ exist separately from the host chromosome. Other phages may enter a ‘chronic’ cycle involving replication separate from the host chromosome and virion production without host lysis. Still other phages exhibit a ‘carrier state’ in which cell lysis in a population is maintained at a low level that does not measurably impact overall population growth [32]. Each of these could lead to extrachromosomal phage sequences appearing in whole-genome shotgun (WGS) metagenomic sequencing data as well as modern draft microbial genome datasets. The presence of these phage data means that PHASTER’s phage prediction routines will have to be adjusted to handle these types of viral signals.
The mining of complete microbial genomes and archived metagenomic sequence data has recently been used to isolate vast amounts of new viral sequence data. This work has led to an increase in the number of known viral sequences and genes by an order of magnitude or more, most of which lack sequence similarity in comparison to previously known phage/viral sequences [32, 33]. Targeted sequencing of phage/viral metagenomes (viromes) from samples of isolated viral-like particles has led to new virome databases such as with Metavir [34] and iVirus [35]. These resources could be better exploited in the next release of PHASTER to help identify previously unknown phages based on new phage sequence data.
Finally, viral metagenome sequencing has also multiplied the number of known viral gene families consisting of auxiliary metabolic genes—viral-encoded host genes used by viruses to metabolically reprogram their hosts during infection—and shown that their biological role is much more extensive than previously thought [36]. We plan to explore how a catalogue of viral auxiliary metabolic genes could be incorporated into PHASTER to enhance its prophage annotation and potentially improve its prophage prediction algorithm’s coverage and accuracy.
Conclusion
PHAST and PHASTER are two important and widely used tools for finding prophages in bacterial genomes. PHASTER, which represents a substantially upgraded version of PHAST, can also be used for finding prophages in contigs collected or assembled in metagenomic studies. In this review, we have given a brief summary of the unique features, strengths and capabilities of both PHAST and PHASTER. We have also provided some practical tips for their use, and discussed some possible areas where they could be improved still further. Tools to automatically find prophages in bacterial genomes can be expected to increase in importance as more and more microbial genomes are sequenced and as more WGS metagenomic surveys are undertaken. As these trends continue, we will continue to work to improve the speed and capabilities of our phage finding tools, resources and algorithms.
Overview of the popular bacterial prophage prediction tool PHAST, its capabilities, features and algorithm.
Enhancements in PHASTER, the successor to PHAST, including improved features and usability, and more efficient data throughput.
Practical guidance is provided on using the PHAST and PHASTER web servers.
Recent developments and potential future improvements to PHASTER are explored.
Funding
The authors wish to thank the Canadian Institutes of Health Research (CIHR) and Genome Alberta, a division of Genome Canada, for financial support.
David Arndt has completed two BSc degrees, one in Molecular Genetics and another in Computing Science. He has worked for 8 years in the Wishart group on bioinformatics projects involving protein structure refinement, metabolomics, phage finding and metagenomics.
Ana Marcu has completed a BSc in Computing Science and Biological Sciences. She has worked in bioinformatics projects involving metabolomics, phage finding and precision health with the Wishart Group and with a precision health company called Molecular You.
Yongjie Liang has a BSc in Biological Sciences from the University of Alberta specializing in bioinformatics. He has worked with the Wishart Group on projects involving protein structure prediction, phage finding and computer parallelization.
David S. Wishart is a professor in the departments of Biological Sciences and Computing Science at the University of Alberta. He has been working in the field of bioinformatics since 1987.

{kind=link}
{kind=link}