





Abstract
Understanding antibody–antigen interactions is key to improving their binding affinities and specificities. While experimental approaches are fundamental for developing new therapeutics, computational methods can provide quick assessment of binding landscapes, guiding experimental design. Despite this, little effort has been devoted to accurately predicting the binding affinity between antibodies and antigens and to develop tailored docking scoring functions for this type of interaction. Here, we developed CSM-AB, a machine learning method capable of predicting antibody–antigen binding affinity by modelling interaction interfaces as graph-based signatures.
CSM-AB outperformed alternative methods achieving a Pearson's correlation of up to 0.64 on blind tests. We also show CSM-AB can accurately rank near-native poses, working effectively as a docking scoring function. We believe CSM-AB will be an invaluable tool to assist in the development of new immunotherapies.
CSM-AB is freely available as a user-friendly web interface and API at http://biosig.unimelb.edu.au/csm_ab/datasets.
Supplementary data are available at Bioinformatics online.
1 Introduction
Antibodies are powerful research and clinical tools, due to their ability to achieve high binding affinity and specificity to antigens. The interactions between antibodies and their targets are mediated by highly variable loops, the complementary determining regions, which have distinctive amino acid propensities considerably enriched in aromatic residues and Asp (Koide et al., 2007), allowing antibodies to form better shape complementarity to diverse epitopes using less close contacts compared with globular protein recognitions. This allows for a small fraction of variable loops to contribute to the majority of antibody–antigen binding energy (Robin et al., 2014), indicating that finding unique features across antibody–antigen binding interfaces should be key to improve the binding affinity and specificity of antibodies.
Structural insights have proven important for guiding antibody design and engineering, but are often limited by availability of experimental structures. Significant efforts have, therefore, been focussed on improving molecular docking approaches (Agrawal et al., 2019); however, their success rate on identification of native poses of antibody–antigen complexes was lower than for enzyme–inhibitor complexes (Yan and Huang, 2019). Most scoring functions also showed only weak correlations to the experimentally determined binding affinities (KD) of antibody–antigen complexes (Guest et al., 2021), highlighting the need for more accurate scoring functions and affinity predictors for antibody design.
Graph-based signatures are a simple way of describing the geometry and physicochemical properties of biological molecules, which allows complex networks of amino acids in antibody–antigen interfaces to be efficiently modelled. We have previously shown that they are a powerful tool for predicting the binding affinity of small molecules to proteins (Pires and Ascher, 2016), and understanding the effects of mutations on antibody binding (Myung et al., 2020a,b; Pires and Ascher, 2016). Here, we have developed CSM-AB, the first dedicated scoring function for antibody–antigen docking and binding affinity predictor by adapting our concept of graph-based signatures in order to capture not only close-contact features but also surrounding structural information relevant for antibody–antigen binding. We show that this is an accurate and scalable way of not only scoring docking poses, but to also predict the antibody binding affinity. We validate CSM-AB using independent blind tests and show it outperforms alternative methods. Details on data collection, feature engineering, model training and evaluation can be found in Supplementary Materials.
2 Results
CSM-AB achieved a Pearson's correlation coefficient of up to 0.40 (RMSE = 1.71 kcal/mol, Supplementary Fig. S1) when attempting to predict antibody–antigen binding affinity on 472 complexes under cross-validation, performance that was consistent for different validation strategies (5-, 10-, 20- and leave-one-out cross validation), demonstrating that there were no significant sampling biases during training. When we compared the performance of our approach with other 18 existing methods, CSM-AB significantly outperformed all of them (P-value < 0.001) when predicting antibody–antigen binding affinity (as the Gibbs Free Energy of binding, ΔG, in kcal/mol) (Supplementary Table S1).
As experimental structures might not always be available, the performance on theoretical models of structures can be important in antibody–antigen docking platforms. To assess the scoring capability of CSM-AB on mutated antibody–antigen complexes, we collected single-point and multiple-point mutants (up to 14 mutations per structure) from mCSM-AB2 (Myung et al., 2020b) and mmCSM-AB (Myung et al., 2020a) datasets, respectively, which were used as blind tests. CSM-AB achieved a Pearson's correlation coefficient of 0.61 (RMSE = 1.68 kcal/mol) and 0.64 (RMSE = 1.75 kcal/mol) on 689 single-point and 301 multiple-point mutations (Supplementary Fig. S2). A significant number of the least accurate predictions (18) were in tight-binders (ΔG = −12 to −14 kcal/mol, for the PDB 3L5X) or (6) of constructs with many point mutations introduced (6 to 14 mutations per case for the PDB 3BDY), which accounted for more than 70% of outliers (Supplementary Fig. S3). Compared with other methods, CSM-AB showed superior performance on both single-point and multiple-point mutants (Supplementary Table S1), significantly outperforming (P-value < 0.001) all 18 methods used in the comparison.
We further investigated the ability of CSM-AB in ranking docked poses on the Dockground (Kundrotas et al., 2018) and ZDOCK benchmark v4 (Hwang et al., 2010) datasets. The predicted ΔG of each complex was ranked and counted if the near-native model was ranked first. CSM-AB showed comparable performance to 18 available tools in scoring docked poses showing six Top1 ranked models (counts of near-native models) out of 15 antibody–antigen complexes (Supplementary Table S1). Also, we measured the performance of available docking methods on local perturbation poses using the ZDOCK dataset which were filtered by CαRMSD and DockQ-CAPRI quality (Basu and Wallner, 2016). Using Kendall's rank correlation coefficient between the predicted ΔG and DockQ-score, CSM-AB ranked Top1 out of 19 methods (Supplementary Table S2) achieving the average Kendall's tau of 0.43. Notably, obtaining correctly docked structures from unbound antibody and antigen chains has been more challenging than of using its bound form due to the possible structural changes upon binding. The 19 tools showed limited performance on the seven antibody–antigen complexes (avg. Kendall’s tau of 0.16) from their unbound forms than the six structures (avg. Kendall’s tau of 0.21) from their original antibody–antigen complexes. CSM-AB also showed better performance on the seven bound complexes (avg. Kendall’s tau of 0.43) than those of the six antibody–antigen complexes (avg. Kendall’s tau of 0.157).
The CSM-AB web-server provides two prediction modes. The single mode predicts the binding affinity of antibody–antigen complexes (ΔG) and allows users to inspect the distribution of interactions found in the antibody–antigen interfaces via bar chart and 3D visualization (Fig. 1). The docking scoring mode allows re-scoring of docked structures. All docked structures should be provided in a single-receptor PDB file and a multiple-pose (up to 100 poses) PDB file. Users can browse the result of each pose in docking scoring mode with the interactive datatable with 3D visualization (Fig. 1). CSM-AB has also been made available as an Application Programming Interface (API) to facilitate incorporation in Bioinformatics pipelines.
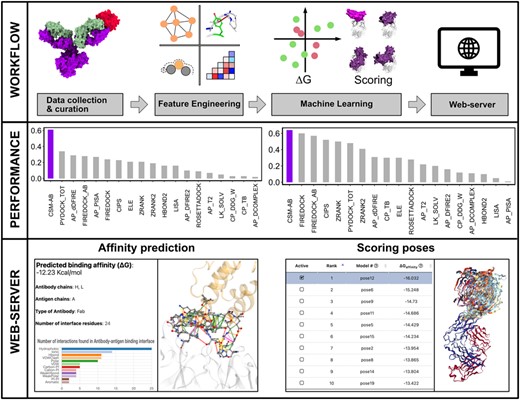
The workflow of this study is described in four steps (top). In data collection and curation, binding affinity datasets (472 training data, 689 single-point mutations and 301 multiple-point mutations) and docking poses (15 Dockground and 13 ZDOCK antibody–antigen complexes) were collected and curated for evaluation of binding affinity and scoring poses. At the feature engineering step, graph-based signatures, Arpeggio interactions, free solvent accessible surface area, residue depth, secondary structure information were extracted and evaluated on collected datasets. During machine learning, all the extracted features were used for cross-validation and greedy feature selection with supervised machine learning algorithms. The performance of CSM-AB and available methods on single-point and multiple-point mutations were compared (middle). On the last step, the best performing model across both binding affinity and scoring docked poses was implemented into CSM-AB webserver. Users can predict ΔG and atomic interactions of an antibody–antigen complex via ‘affinity prediction’ mode and re-score up to 100 docked poses via ‘scoring poses’ mode (bottom)
3 Conclusions
Antibody–antigen docking approaches have been widely needed for accelerating lead characterization and optimization processes of immunotherapy development. While significant effort has been devoted to improve the performance of docking scoring functions, currently available methods show limited accuracy in predicting the binding affinity of antibody–antigen complexes.
CSM-AB is a machine learning approach, taking account of structural features such as graph-based signature and atomic interactions across the antibody–antigen interface residues. Compared to other approaches, CSM-AB presented better performance on cross-validation and blind test, sets demonstrating its capability of binding affinity prediction and applicability in re-scoring docked antibody–antigen complexes. CSM-AB is available as a user-friendly web-server and API at http://biosig.unimelb.edu.au/csm_ab/.
Funding
This work was supported by an Investigator Grant from the National Health and Medical Research Council (NHMRC) of Australia [GNT1174405 to D.B.A.] and the Victorian Government's OIS Program. Y.M. was supported by the Melbourne Research Scholarship.
Conflict of Interest: none declared.


{kind=link}