





Abstract
Histones form octameric complexes called nucleosomes, which organize the genomic DNA of eukaryotes into chromatin. Each nucleosome comprises two copies each of the histones H2A, H2B, H3 and H4, which share a common ancestry. Although histones were initially thought to be a eukaryotic innovation, the subsequent identification of archaeal homologs led to the notion that histones emerged before the divergence of archaea and eukaryotes.
Here, we report the detection and classification of two new groups of histone homologs, which are present in both archaea and bacteria. Proteins in one group consist of two histone subunits welded into single-chain pseudodimers, whereas in the other they resemble eukaryotic core histone subunits and show sequence patterns characteristic of DNA binding. The sequences come from a broad spectrum of deeply-branching lineages, excluding their genesis by horizontal gene transfer. Our results extend the origin of histones to the last universal common ancestor.
Supplementary data are available at Bioinformatics online.
1 Introduction
Eukaryotes condense their genomic DNA into a highly repetitive structure called chromatin. The basic unit of chromatin, the nucleosome, is evolutionarily conserved in eukaryotes, comprising 147 base pairs of double-stranded DNA, wrapped around an octameric core complex (Arents et al., 1991; Kornberg and Thomas, 1974; Luger et al., 1997). In this core complex, two copies of each of the four histone proteins H2A, H2B, H3 and H4 are organized as a central (H3–H4)2 tetramer flanked by two H2A–H2B dimers (Arents et al., 1991; Luger et al., 1997). The different subunits exhibit pairwise sequence similarity indicative of a common ancestry. They assume a common fold, featuring three helices separated by two short strap loops, which are organized into two consecutive helix-strand-helix (HSH) motifs (Arents et al., 1991). The core histones contain additional N- and/or C-terminal extensions (the histone tails), which play a role in gene regulation and chromatin assembly (Jenuwein and Allis, 2001; Luger and Richmond, 1998).
The histone fold is also found in several other widespread eukaryotic proteins, mostly involved in DNA metabolism; examples include the B and C subunits of the nuclear transcription factor Y (NF-YB3, NF-YC2), the TATA box-binding protein-associated factors (TAFs), the chromatin accessibility complex proteins CHRAC14 and CHRAC16 as well as the centromere proteins CENP-S, CENP-T, CENP-W and CENP-X. (Arents and Moudrianakis, 1995; Gnesutta et al., 2013). A variant form is exhibited by the histone-fold domain of the Son of Sevenless protein (SOS), in which the subunits are located consecutively on a single polypeptide chain to yield a pseudodimer (Sondermann et al., 2003).
Histones have historically been thought to be hallmarks of eukaryotes. However, over the last several years, homologs that form higher-order complexes with DNA have been identified in most archaeal lineages (Ammar et al., 2012; Mattiroli et al., 2017; Pereira et al., 1997; Reeve et al., 1997; Sandman and Reeve, 2000, 2006). Archaea typically possess only one or two histones of the HMfB-like family, composed of merely the histone fold (Sandman and Reeve, 2006). These assemble into homodimers (e.g. HMfA and HMfB homodimers of Methanothermus fervidus) or, in some cases, possess a variant with two subunits on a single polypeptide chain, similar to the form seen in the aforementioned SOS protein (e.g. HMk of Methanococcus kandleri) (Fahrner et al., 2001). Unlike eukaryotic core histones, which always assemble into discrete octameric particles, archaeal HMfB-like histones form extended structures with variable numbers of dimers. These wrap DNA in a quasi-continuous superhelix, which geometrically resembles eukaryotic nucleosomal DNA (Bhattacharyya et al., 2018; Maruyama et al., 2013; Mattiroli et al., 2017). The formation of such extended structures has been shown to be important for gene regulation in archaea (Mattiroli et al., 2017).
Bacteria have generally been thought to lack histone homologs. However, structures of two bacterial histone-fold proteins, exhibiting two subunits on a single chain, have been reported. These proteins originate from Aquifex aeolicus [Aq_328; Protein Data Bank (PDB) 1R4V] and Thermus thermophlius (TTHA1479; 1WWI), with reported homologs in just a few other bacteria (Qiu et al., 2006). They are classified into the same homology group as the other histone-fold proteins in the Structural Classification of Proteins extended (SCOPe) (Fox et al., 2014) and Evolutionary Classification of protein Domains (ECOD) (Cheng et al., 2014) classification systems, and into the DUF1931 family in the Pfam domain database (Finn et al., 2016). Because of this seeming absence of histone homologs in most bacteria and their presence in most archaea, it is presently thought that the histone fold originated before the divergence of archaea and eukaryotes, and that it subsequently radiated to some bacteria through horizontal gene transfer (Alva et al., 2007; Sandman and Reeve, 2006).
In a previous study, aimed at obtaining further insight into the origin of the histone fold, we found evidence for its common origin with the helical part of the extended AAA+ ATPase domain (C-domain) based on the presence of a shared primordial HSH motif (Alva et al., 2007). We hypothesized that this motif gave rise divergently to the AAA+ C-domain as well as to a further fold, the N-domain of Clp/Hsp100 proteins, and that the histone fold arose subsequently from the C-domain through a three-dimensional domain-swapping event; this latter point has received experimental support (Hadjithomas and Moudrianakis, 2011). Since the AAA+ ATPase domain is ubiquitous to life, it must have existed already at the time of the last universal common ancestor (LUCA), but exactly when the histone fold may have emerged is still unclear. Also, although structures of the two aforementioned bacterial histone-fold proteins have been in the PDB for ∼15 years, systematic searches for histone homologs in bacteria are lacking. Here, we describe the detection of two further prokaryotic histone-like groups, with presence in many deep-branching bacteria as well as in archaea. One of these is exemplified by the bacterial single-chain pseudodimers Aq_328 and TTHA1479, whereas the other resembles eukaryotic core histones and archaeal HMfB-like histones in containing just one copy of the histone fold.
2 Materials and methods
HHpred searches were carried out in the MPI Bioinformatics Toolkit (Soding, 2005; Zimmermann et al., 2018) against the PDB_mmCIF70, SCOPe70 and ECOD_F70 databases, as well as against the profile HMM databases of genomes of many phylogenetically diverse bacteria. We switched off secondary structure scoring to eliminate the possibility that the matches were scored highly because of a chance similarity of their secondary structures. Three iterations of HHblits (Remmert et al., 2011) against the Uniclust30 database (Mirdita et al., 2017), with an E-value inclusion threshold of 1e−3, were used for building profile HMMs. Searches were carried out with representatives of histone-fold proteins as well as of their distant homologs, chosen from SCOPe (Fox et al., 2014) and ECOD (Cheng et al., 2014) to cover all histone-fold families of known structure (the representatives are provided in the Supplementary Material S1).
To identify sequences for cluster analysis, we searched the non-redundant (nr) protein sequence database at NCBI for homologs of the aforementioned representatives, using two iterations of JackHMMER in default settings (Johnson et al., 2010). We pooled together the obtained sequences and filtered them down to a maximum pairwise sequence identity of 70% using CD-HIT in default settings (Huang et al., 2010). The sequences in the filtered set were clustered in CLANS (Frickey and Lupas, 2004) based on the strength of their all-against-all pairwise P-values, obtained by running two iterations of JackHMMER. Clustering was done to equilibrium in 2D at a P-value cutoff of 1e−07 using default settings, except for attract value =1 and repulse value =30.
3 Results and discussion
We used sensitive sequence comparisons, as implemented in HHpred, to investigate evolutionary relationships of the two aforementioned bacterial histone-fold pseudodimers, Aq_328 and TTHA1479, to archaeal HMfB-like and eukaryotic histone proteins. We built separate profile HMMs for the N- and C-terminal halves of the bacterial pseudodimers and compared them with the profile HMMs of several representative histone-fold proteins, including the nucleosome core histones, the two halves of the SOS histone domain, the histone domains of TATA box-binding protein-associated factors, archaeal histones and AAA+ C-domains (Fig. 1). The N-terminal halves made many statistically significant connections that are indicative of homology to the archaeal and eukaryotic proteins, with HHpred probability values of >90%. While their best matches were to the archaeal histones, their most distant matches were to the AAA+ C-domains and ClpA N-domains. Curiously, however, the N-terminal halves made no significant connections to the C-terminal ones. Conforming to this, reciprocal searches with the C-terminal halves did not yield any significant matches, either to the N-terminal ones or indeed to any other histone-fold protein, suggesting that they represent a highly divergent form of the histone fold. Additional HHpred searches against SCOPe70, ECOD70 and PDB70 also did not return any significant matches. In contrast to this, the two halves of the eukaryotic SOS and archaeal HMk single-chain variants exhibited clear sequence similarity to each other.
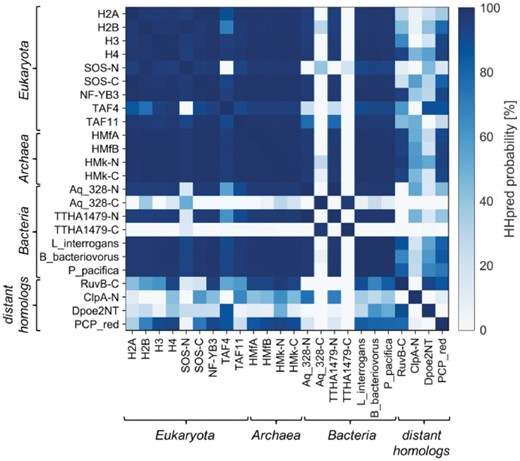
Pairwise HMM comparison of histone-fold proteins. The N- and C-terminal domains of the SOS, HMk, Aq_328 and TTHA1479 pseudodimers were considered separately in the comparisons. Accession information for the proteins is provided in the Supplementary Material S1 and a gallery of structures in Supplementary Figure S1
To detect further, possibly divergent homologs of the bacterial histone-fold proteins, we searched PDB70 as well as databases of several representative bacterial genomes. For this, we used HHpred seeded with the N-terminal half of Aq_328, the archaeal histone HMfB and the four eukaryotic core histones. Although the searches against PDB70 found no further bacterial proteins of the histone fold, they identified many homologous connections, with HHpred probability values of >70% (Fig. 1), to domains of two structurally distinct folds, which represent additional embodiments of the aforementioned HSH motif: the N-terminal domain of the B subunit of human DNA polymerase epsilon (Dpoe2NT; PDB 2V6Z) (Nuutinen et al., 2008) and the PCP_red domain of the B subunit of bacterial light-independent protochlorophyllide reductase (PDB 2KRU). Topologically, these proteins resemble the C-domain of AAA+ proteins; however, PCP_red appears to have lost the C-terminal helix, as some C-domains have done as well (Ammelburg et al., 2006). In the ECOD database, both proteins are grouped within the Histone-related proteins. HHpred searches against bacterial genomes yielded additional homologs of the bacterial pseudodimer from Frankia alni, Nostoc punctiforme and Thermus aquaticus as well as hitherto undescribed homologs, comprising just one copy of the histone fold, from Bacteriovorax sp. DB6 IX, Bdellovibrio bacteriovorus, Leptospira interrogans, Plesiocystis pacifica, Streptomyces scabiei and Waddlia chondrophila. Reciprocal searches with these proteins against several representative histone-fold domains confirmed their membership in the histone superfamily. Like the single-chain pseudodimers, the bacterial homologs with a single copy of the histone fold also made their best matches to the archaeal histones (Fig. 1).
Having established the presence of two types of histone-fold proteins in bacteria, one comprising one copy of the domain and the other two, we explored their preponderance and phylogenetic distribution in bacterial species by searching for homologs in the nr protein sequence database using JackHMMER. The search seeded with Aq_328 yielded about 360 bacterial homologs, almost all of which contain two consecutive copies of the histone fold. While around 70% of these homologs come from the deep-branching, ancient class Actinobacteria, the rest are scattered sparsely across several diverse classes, including Aquificae, Bacteroidetes, Chloroflexi, Cyanobacteria, Deinococcus and Firmicutes as well as most classes of Proteobacteria. Some organisms contain two homologs of the pseudodimer; for instance, A. aeolicus, in addition to Aq_328, possesses a second copy in Aq_616. We also found about 25 homologs in archaea, primarily in organisms of the phylum Euryarchaeota, which additionally also contain members of the archaeal HMfB-like histones. For example, the archaeon Archaeoglobus profundus contains one homolog of the pseudodimer and two of the archaeal HMfB-like histone. This broad occurrence of the pseudodimeric histone-fold proteins in diverse bacteria, including many deep-branching classes, as well as their occurrence in archaea, excludes the role of horizontal transfer in their emergence and suggests that they arose before the divergence of the bacterial and archaeal lineages.
The search seeded with a representative of the second bacterial type, composed of a single histone fold, from the bacterium L. interrogans, returned about 230 bacterial homologs from Deltaproteobacteria, Oligoflexia, Spirochaetes, Planctomycetes, Elusimicrobia, Chlamydiae as well as some from the Parcubacteria group. While the majority of these are 60–70 residues long and consist of just the histone fold, in which the C-terminal α-helix is seemingly shorter, some contain two consecutive, homologous copies of the histone fold or an additional N-terminal zinc-binding domain. Some bacteria contain multiple copies of this type, but we found no instances where they co-occur with the pseudodimeric type. Also, in contrast to the pseudodimer, these proteins are more widespread in archaea, with about 500 homologs distributed across most lineages, including the Asgard group, which has been proposed to represent the closest prokaryotic relative of eukaryotes (Spang et al., 2015). They frequently co-occur with archaeal HMfB-like histones and occasionally also with the pseudodimeric type. The extremophilic archaeon Pyrococcus furiosus, for instance, contains two archaeal HMfB-like histones and one homolog each of the two bacterial groups. The broad distribution of this second type of histone-fold proteins in bacteria as well as in archaea suggests that it also emerged before the divergence of archaea and bacteria.
To confirm the sequence relationships between histone-fold proteins on a global scale, we gathered their homologs from the nr protein sequence database and clustered them in CLANS based on the strength of their all-against-all pairwise sequence similarities. CLANS, an implementation of the Fruchterman–Reingold force-directed layout algorithm, treats protein sequences as point masses in a virtual multidimensional space, in which they attract or repel each other based on the strength of their pairwise similarities. Consequently, evolutionarily related sequences gravitate to same parts of the map, forming clusters of related sequences. In the obtained map (Fig. 2), the archaeal HMfB-like histones, including the HMk-like pseudodimers, form a central cluster. The various eukaryotic clusters are tightly connected to it, highlighting their evolutionary emergence and radiation from a shared prototype that existed before the divergence of the archaeal and eukaryotic lineages. We also found histone homologs from viruses in the map, all of which lie within the clusters of the eukaryotic core histones, indicating that they were acquired by horizontal transfer from eukaryotes. The two types of bacterial histone-fold proteins and their respective archaeal homologs, however, form two separate clusters, distinct from the core archaeal and eukaryotic clusters.
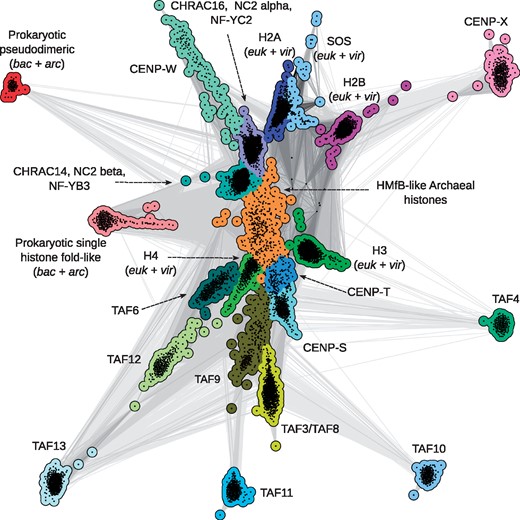
Cluster map of the histone superfamily. Homologs of archaeal, bacterial and eukaryotic histones were gathered from the nr protein sequence database, filtered to 70% identity and clustered in CLANS. Dots represent sequences and those within the same group are shown in one color. Line coloring reflects the significance of sequence similarities; the darker a line, the higher the significance. Accession information for representatives of all clusters are provided in Supplementary Material S1
An analysis of sequence patterns important for DNA binding in eukaryotic core histones (the RT-pair and the RD-clamp; Fig. 3) shows that these are widely represented in the group of archaeal HMfB-like proteins (Mattiroli et al., 2017), which gave rise to the eukaryotic nucleosome subunits and themselves form higher-order complexes with DNA (Mattiroli et al., 2017; Sandman and Reeve, 2006). They are almost equally widely represented in the prokaryotic single histone fold-like group, raising the possibility that the proto-histone fold at the time of the LUCA was already a DNA binding protein. The sequence patterns characteristic for DNA-binding are largely absent from the prokaryotic pseudodimeric group, in which as aforementioned only the N-terminal halves are homologous to the other histone-fold proteins. We note that the N-terminal halves of the bacterial pseudodimers predominantly possess the RD-clamp. This general absence of DNA-binding motifs in the prokaryotic pseudodimers, however, does not contradict the ancestrality of DNA-binding, as this form is most likely derived from a dimeric form and other such derived pseudodimers, such as the histone domain of SOS, are known to have lost DNA binding activity.

Conservation of DNA-binding residues in histones. Amino acid residues part of the conserved RT-pair and RD-clamp, which are important for interactions with DNA both in eukaryotic and archaeal DNA-histone complexes, are indicated by a colon (:) and pipe (|), respectively. The secondary structure below the alignment (h=α-helix, b=β-strand) is that of the experimentally determined structure of human H2A protein. For the prokaryotic pseudodimers, sequences of only the N-terminal halves are shown; the C-terminal halves were excluded as they do not make homologous connections to any other histone-fold protein. Accession information for the sequences is provided in the Supplementary Material S2
4 Conclusion
In summary, we identified two further prokaryotic groups of histone-fold proteins, single histone fold-like and pseudodimeric groups, both with a broad but sparse distribution in many bacterial and archaeal lineages, indicating that the histone fold was already established at the time of the LUCA. We also describe two further deep homologs of histone-like proteins, the N-terminal domain of the B subunit of human DNA polymerase epsilon (Dpoe2NT) and the PCP_red domain of the B subunit of bacterial light-independent protochlorophyllide reductase, both showing the same topology as the C-domains of AAA+ proteins, lending additional support to our previous proposal that a protein of this type gave rise to the proto-histone fold by 3D domain-swapping and dimerization (Alva et al., 2007). Thus, in this scenario, the first histone-like fold was a homodimer. The subsequent differentiation of the fold variously yielded heterodimeric forms as well as fused, pseudodimeric ones on multiple, independent occasions. A key question is at which point the ability to bind DNA arose. The wide representation of patterns important for DNA binding in the prokaryotic single histone fold-like group suggests that the proto-histone at the time of the LUCA had already established the ability to interact with DNA. Correspondingly, we propose that the bacterial single histone fold-like proteins cause a similar wrapping of bound DNA as the archaeal and eukaryotic histones. DNA binding activity may even have pre dated the emergence of the histone fold, since the fold from which it arose, represented today by the C-domain of AAA+ proteins, is also often associated with DNA binding (Alva et al., 2007; Lin et al., 2009; Nuutinen et al., 2008). Even the fragment whose duplication gave rise to the C-domain itself is thought to have originated in an ancient RNA-peptide world, where it could have been involved in interactions with nucleic acids (Alva et al., 2007; Alva et al., 2015).
Funding
This work was supported by institutional funds from the Max Planck Society.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}