





Abstract
Somatic Mutation calling method using a Random Forest (SMuRF) integrates predictions and auxiliary features from multiple somatic mutation callers using a supervised machine learning approach. SMuRF is trained on community-curated matched tumor and normal whole genome sequencing data. SMuRF predicts both SNVs and indels with high accuracy in genome or exome-level sequencing data. Furthermore, the method is robust across multiple tested cancer types and predicts low allele frequency variants with high accuracy. In contrast to existing ensemble-based somatic mutation calling approaches, SMuRF works out-of-the-box and is orders of magnitudes faster.
The method is implemented in R and available at https://github.com/skandlab/SMuRF. SMuRF operates as an add-on to the community-developed bcbio-nextgen somatic variant calling pipeline.
Supplementary data are available at Bioinformatics online.
1 Introduction
Identification of somatic mutations from matched tumor and normal samples is challenged by sequencing noise and alignment ambiguities as well as the heterogeneous composition of tumors. Recent studies have revealed low concordance between existing methods for somatic variant calling (Hwang et al., 2015; Kroigard et al., 2016; O’Rawe et al., 2013; Roberts et al., 2013). Additionally, a benchmark study demonstrated that the accuracy of a given somatic mutation calling algorithm can vary extensively across different workflows and pipelines (Alioto et al., 2015). Parameters influencing this variation may be choice of alignment algorithm, use of local re-alignment, as well as configuration of a multitude of post-processing filters. The consensus of multiple callers have been used to improve the accuracy of somatic variant calling (Callari et al., 2017; Ellrott et al., 2018; Rashid et al., 2013). Taking this one step further, a machine learning based ensemble method may combine multiple mutation callers with auxiliary sequence and alignment features to improve mutation calling accuracy (Ding et al., 2012; Fang et al., 2015; Wood et al., 2018). While such approaches may improve accuracy, they are generally not portable: The end-user must obtain suitable training and testing datasets and need to have knowledge of machine learning (Supplementary Fig. S1A ). There is therefore a need for accurate and pre-trained ensemble approaches for somatic mutation calling that can be ported between research groups. Here, we developed a Somatic Mutation calling method using a Random Forest (SMuRF), which combines predictions from four mutation callers with auxiliary alignment and mutation features using supervised machine learning (Supplementary Fig. S1B).
2 Implementation
SMuRF is available as an R package. Briefly, the bcbio-nextgen framework (https://github.com/chapmanb/bcbio-nextgen) is used to generate somatic variant calls from 4 different methods: MuTect2 (Cibulskis et al., 2013), Freebayes somatic (ArXiv: https://arxiv.org/abs/1207.3907), VarDict (Lai et al., 2016) and VarScan (Koboldt et al., 2012). Variant and auxiliary features are extracted from the VCF files. The SMuRF random forest model is pre-trained on a gold standard set of mutation calls curated by the International Cancer Genome Consortium (ICGC) community using deep (>100×) whole genome sequencing (WGS) of two tumors (Alioto et al., 2015). Feature extraction and prediction of somatic variants takes ∼10 min for tumor-normal WGS data on a standard computer (4 CPUs, 16GB RAM).
3 Overview
SMuRF SNV and indel models were trained on matched tumor-normal WGS data from a chronic lymphocytic leukemia (CLL) patient and a medulloblastoma (MB) patient, where the true somatic mutations have been identified and curated by the International Cancer Genome Consortium (ICGC) (Alioto et al., 2015). The training data was augmented to expose the model to additional variation in sequencing coverage, tumor purity and tumor/normal coverage imbalance (Supplementary Fig. S1C and Supplementary Methods). SMuRF was trained on 80% of the data, with 20% of the data withheld as a test set. Highly predictive features were mostly somatic variant scores provided by individual methods as well as mapping and base quality estimates (Supplementary Table S1). SMuRF achieved F1-scores of 0.88 and 0.74 for SNVs and indels, respectively (Fig. 1A and B, Supplementary Tables S2 and S3 and Supplementary Fig. S3 for SNV coding regions). Importantly, SMuRF showed improved accuracy over the best mutation calling submissions reported in the benchmark by Alioto et al. using the same dataset (best reported F1-scores 0.79 and 0.65 for SNV and indels, respectively) (Alioto et al., 2015). In our analysis, while individual methods could recover most of the true SNVs, this came at the cost of very low precision (<40% precision at 80% recall) (Supplementary Table S2). In contrast, SMuRF could recover 86% of the true SNVs (recall) at 92% precision on the withheld test set. While a simple consensus approach using the intersection of individual methods performed well (F1 = 0.82), SMuRF achieved markedly higher recall (86% versus 74%) at a similar level of precision. All methods, including SMuRF, were mostly robust when tested under different levels of tumor purity (Supplementary Fig. S4). However, SMuRF showed substantially improved accuracy at low somatic variant allele frequencies (VAFs) as compared to individual methods (Fig. 1C), which is particularly important in the setting of tumor heterogeneity inference (Shi et al., 2018). We further benchmarked SMuRF SNV calling using independent data from the DREAM Somatic Mutation Challenge where artificial tumor data has been generated using an in-silico approach (Ewing et al., 2015). While the performance of individual methods varied across these datasets, SMuRF was highly accurate across all synthetic tumors (F1 > 0.8) (Supplementary Fig. S5). Overall, these results support that SMuRF is robust and can generalize to unseen data.
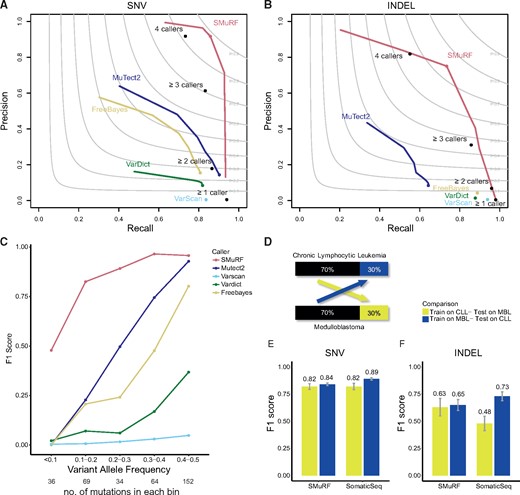
Performance of SMuRF. Precision-recall profiles for individual somatic mutation callers and SMuRF evaluated on (A) SNV and (B) indels using 20% withheld test data. Curves show the performance of the individual algorithms under different variant score thresholds (MuTect2 tumor log-odds score, Freebayes log-odds score, VarDict SSF score, VarScan SSC score and SMuRF confidence score). Solid points refer to the default performance of the caller in the bcbio-nextgen workflow. Black solid points denote the accuracy of calls identified by the majority-voting scheme in bcbio-nextgen (at least 1, 2, 3 or 4 callers). The grey contours indicate F1 scores as a function of recall and precision. (C) Accuracy of SMuRF and individual callers as a function of somatic variant allele frequency in the test set; F1 scores evaluated for each variant allele frequency bin. (D–F) Evaluation of SMuRF and SomaticSeq performance when trained and tested across different cancer types. (D) Models were trained on 70% of CLL data and tested on 30% of MB data (and vice versa). F1 scores were recorded for SMuRF and SomaticSeq SNV (E) and indel (F) predictions. Error bars represent the standard deviation of the mean across 10 random training/test data splits (same splits for both methods)
Analysis of indel prediction accuracy showed that individual mutation callers could recover most of the true indels (64–94% recall), but only at the cost of very low precision (<8%). Interestingly, simple consensus approaches performed well for indel prediction (F1 0.46 and 0.66 for 3 and 4-caller consensus, respectively). However, while consensus methods suffered from either low recall (0.55) or precision (0.31), SMuRF obtained high indel prediction accuracy (F1 = 0.74) with both high recall (0.74) and precision (0.75) (Fig. 1B, Supplementary Table S3). We also analyzed the extent that SMuRF predicts the same somatic mutations in tumor samples profiled with both (>200× coverage) WES and (<100× coverage) WGS. When restricting analysis to variants in coding regions, SMuRF predicted somatic SNVs and indels with comparable or higher concordance than individual methods (Supplementary Figs S6 and S7).
Finally, we compared SMuRF to two existing machine learning-based methods. The first was MutationSeq, a pre-trained ensemble SNV caller (Ding et al., 2012), which achieved an F1-score of 0.68, similar to the other individual SNV callers in our analysis (Supplementary Fig. S8). Next, we compared the performance of SomaticSeq (Fang et al., 2015), a method that required users to train their own predictive model (see Supplementary Methods). The trained SomaticSeq model had slightly increased test set prediction accuracy over SMuRF for both SNV (0.90 versus 0.88) and indels (0.78 versus 0.75) (Supplementary Fig. S9B and C). We further evaluated how the methods generalized when models were trained and tested across different tumor datasets and found that SomaticSeq showed greater test accuracy variation (Fig. 1D–F). This was especially pronounced for indel prediction, where the F1 accuracy of SomaticSeq varied from 0.48 to 0.73 (SMuRF 0.63–0.65) when tested on the MBL or CLL sample, respectively. Furthermore, SomaticSeq used ∼24 h to predict both SNVs and indels since it also computes auxiliary features from the raw alignment data. In contrast, SMuRF depends only on VCF files and predicts both SNVs and indels in ∼10 min (Supplementary Fig. S9A). Overall, these results support that SMuRF is both accurate and computationally efficient.
In summary, SMuRF is an accurate, portable and user-friendly ensemble-based somatic mutation caller, which should benefit both cancer genomics studies as well as clinical applications.
Acknowledgements
We thank I. Kassam, N. Rohatgi, K. Krishnamachari, M. N. Mojtabavi, G. Zhu, U. Ghoshdastider and T. Kulshrestha for their support and discussion during the development and testing of SMuRF.
Funding
This work was supported by an Open Fund Individual Research Grant from the Singapore National Medical Research Council (OFIRG15nov072).
Conflict of Interest: none declared.
References


{kind=link}