





Abstract
A lack of accurate computational tools to guide rational mutagenesis has made affinity maturation a recurrent challenge in antibody (Ab) development. We previously showed that graph-based signatures can be used to predict the effects of mutations on Ab binding affinity.
Here we present an updated and refined version of this approach, mCSM-AB2, capable of accurately modelling the effects of mutations on Ab–antigen binding affinity, through the inclusion of evolutionary and energetic terms. Using a new and expanded database of over 1800 mutations with experimental binding measurements and structural information, mCSM-AB2 achieved a Pearson’s correlation of 0.73 and 0.77 across training and blind tests, respectively, outperforming available methods currently used for rational Ab engineering.
mCSM-AB2 is available as a user-friendly and freely accessible web server providing rapid analysis of both individual mutations or the entire binding interface to guide rational antibody affinity maturation at http://biosig.unimelb.edu.au/mcsm_ab2
Supplementary data are available at Bioinformatics online.
1 Introduction
Antibodies (Abs) are central components of our immune system that bind specifically to their target antigens in order to elicit an immune response. This interaction between an Ab and its antigen is mediated by a myriad of non-covalent interactions made by the complementary determining regions (CDRs) of the Abs with a specific epitope on an antigen. This ability to bind to a wide variety of targets, including those traditionally considered undruggable, in a highly specific and selective manner has led to increasing interest in their use as therapeutics for a broad range of diseases including several types of cancer (Elgundi et al., 2017) and rheumatoid arthritis (Tanaka et al., 2014). Since the first approval of monoclonal Ab, the significant improvement in Ab engineering has led Abs to become best-selling drugs accounting for over half of the therapeutic market (Urquhart, 2018).
Ab development often requires optimization of its stability, solubility, selectivity, affinity and immunogenicity. Achieving the desired properties can often become a major challenge, considering the large number of possible variations in Abs, and with each potentially affecting multiple biological properties. One of the early steps in the development of effective Ab therapies is the engineering of binding specificities and selectivities, which has traditionally been inspired by the natural biological process of affinity maturation, with rounds of mutations within the CDR loops explored. This process can be time-consuming, and is inherently a random and error-prone process. Recent examples, however, have shown how the computationally guided rational engineering of Ab-binding affinities can dramatically improve this process (Kiyoshi et al., 2014; Sefid et al., 2019).
A number of different computational approaches which use an available crystal structure to guide Ab design and optimization have been developed (Roy et al., 2017). A systematic evaluation of the accuracy of these approaches to predict the change upon mutation in binding affinity highlighted the limited performance of existing tools, and the challenging nature of this problem.
In a previous work, we have adapted the concept of graph-based signatures which can efficiently represent the physicochemical properties and geometry of surrounding environment of the wild-type and mutant residues to accurately predict the effects of mutations in terms of protein stability (Pandurangan et al., 2017a; Pires et al., 2014a, b; Rodrigues et al., 2018b) and interactions with other proteins (Pires et al., 2014b; Rodrigues et al., 2019), nucleic acids (Pires et al., 2014b; Pires and Ascher, 2017), small molecules (Pires et al., 2015; Pires and Ascher, 2016a) and metal ions (Pires et al., 2016b). These have been successfully used to provide valuable insights into genetic diseases (Albanaz et al., 2017; Andrews et al., 2018; Ascher et al., 2019; Casey et al., 2017; Hnizda et al., 2018; Jafri et al., 2015; Jubb et al., 2017; Nemethova et al., 2016; Pandurangan et al., 2017b; Ramdzan et al., 2017; Rodrigues et al., 2018a; Silvino et al., 2016; Soardi et al., 2017; Traynelis et al., 2017; Trezza et al., 2017; Usher et al., 2015), drug resistance (Ascher et al., 2015; Hawkey et al., 2018; Holt et al., 2018; Karmakar et al., 2018, 2019; Phelan et al., 2016; Pires et al., 2016a, b; Portelli et al., 2018; Vedithi et al., 2018) and rational protein engineering. We have also successfully applied our graph-based signatures to the prediction of changes in Ab–antigen binding affinity and showed that this outperformed existing methods, although there was still significant room for improvement (Pires and Ascher, 2016b). The release of SKEMPI2.0 containing information of the effects of new mutations on Ab–antigen binding affinity, allowed us to not only assess earlier approaches based on new unseen experimental data, but to also build a predictive model across a more comprehensive set of Ab–antigen complexes and mutations. In particular, mCSM-AB only considered structural information, however evolutionary information and energetic terms have been shown to help predict the effect of a mutation on Ab-binding affinity, as variants which have destabilizing effects on proteins are less likely to be conserved from an evolutionary perspective (Gonzalez-Munoz et al., 2012). In addition, Ab–antigen interfaces are enriched with specific type of amino acids such as Tyr and Ser (Jubb et al., 2015; Van Regenmortel, 2014) compared with other protein–protein complexes, and different modes of interatomic interaction may be important to explain whether the mutation is favourable in its surroundings.
A powerful and scalable model for predicting the effects of missense mutations on Ab-binding affinity could hold enormous potential for guiding rational Ab development. Here we introduce mCSM-AB2, an updated and optimized version of our previous method, trained on a larger and more comprehensive dataset, which uses not only graph-based signatures but also interatomic interaction, evolutionary and energy-based features to capture additional structural and sequence-based information to more accurately predict Ab–antigen affinity changes upon mutation. We show that mCSM-AB2 significantly outperforms existing methods, and has potential to guide rational Ab engineering.
2 Materials and methods
The general mCSM-AB2 workflow is depicted in Figure 1. It is composed of three main steps including: (i) dataset acquisition, which refers to collecting experimental evidence from the literature on effects of mutations in Ab–antigen binding affinity complexes with solved structures; (ii) feature engineering, which encompasses the generation and evaluation of features selected to model different aspects involved in Ab–antigen recognition and effects of mutations on these complexes and (iii) machine learning, which aims to train, test and validate an accurate predictive model via supervised learning, using the computed features and experimental effects of mutations, as evidence.
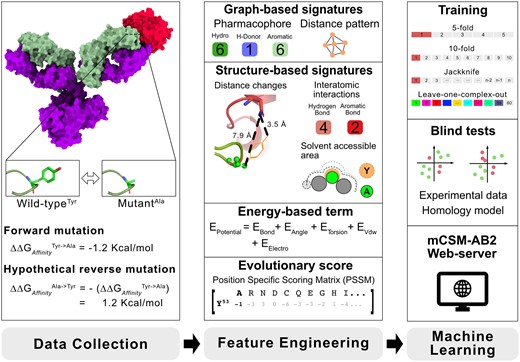
Overview of the mCSM-AB2 workflow. In data collection, after data acquisition, hypothetical reverse mutations are considered to avoid the natural bias of mutations reducing affinity. From the complete dataset (1810 mutations), a range of different features are calculated to be used as evidence to train predictive models using machine learning algorithms. Among the feature classes, graph-based signatures are used to describe the wild-type residue environment and its geometry and physicochemical properties. Other structural attributes aiming to model other relevant aspects driving Ab-antigen affinity were also considered, including the variation in the distance to the antigen upon mutations, solvent accessible area, as well as energetic terms and interatomic interactions. Additionally, an evolutionary score (derived from PSSM) was also used to model mutation tolerance throughout evolution. All features are then used to build predictive models through a series of training and blind-test validation procedures. The best model was then made available as an easy-to-use web server at http://biosig.unimelb.edu.au/mcsm_ab2
2.1 Datasets
To develop our predictive model, we collected binding affinity data with experimentally determined structures from the AB-BIND (Sirin et al., 2016), PROXiMATE (Yugandhar et al., 2017) and SKEMPI2.0 (Jankauskaite et al., 2018) databases to train and test mCSM-AB2. Compared with the earlier AB-BIND dataset used for mCSM-AB, we discarded 3 redundant mutations and 27 mutant non-binders, which led to a dataset of 558 mutations that was used as the training set. SKEMPI2.0 contained 830 single point mutations from Ab–antigen complexes, which were filtered using ‘Hold_out_type’ and ‘Hold_out_proteins’ information, to avoid redundancy during training. Of these 830 instances, there were 102 mutations that had more than one experimentally measured binding affinity for the same mutation, for which we preferentially kept direct binding assays such as SPR or ITC, leading to a group of 728 mutations. Within the 728 mutations selected from SKEMPI2.0, we also disregarded 32 mutant non-binders, leaving a total of 696 mutations in Ab–antigen complexes. Comparing to AB-BIND, our filtered SKEMPI2.0 dataset contained 377 new mutations in unique complexes not present in the original training set. CD-HIT (Huang et al., 2010) was used to cluster interfaces with a similarity threshold of 90%. The new SKEMPI2.0 dataset contained unique interfaces not present in the original AB-BLIND dataset, and was hence used as a non-redundant blind test and comparative tool to evaluate previous methods built using AB-BIND. In total, our train/blind-test datasets are composed of 905 single mutations and the mutations from 11 out of 60 Ab–antigen complexes are present in both training (AB-BIND) and blind test sets (Supplementary Table S1). This represents not only a significant increase over the 558 mutations used to train mCSM-AB spanning across more than twice the number of 25 Ab-antigen complexes, but also a non-redundant experimental blind test set (377 mutations) which allows us to explicitly compare the performance of our new approach to mCSM-AB and other methods.
Due to the nature of experimental affinity maturation, these datasets were unbalanced, with 652 destabilizing (ΔΔGAffinity< 0), 196 stabilizing (ΔΔGAffinity> 0) and 57 neutral (ΔΔGAffinity= 0) single point mutations (Supplementary Figure S1, left). As has been previously proposed (Thiltgen and Goldstein, 2012), to avoid any subsequent bias in our predictive model we also considered the hypothetical reverse mutations, using mutant structures generated by FoldX (Eswar et al., 2006). This gave a final dataset of 1810 single point mutations (Supplementary Figure S1, right), of which 1056 were used for training, and a non-redundant set of 754 mutations were used as a blind test set to avoid overtraining and to benchmark the performance of mCSM-AB2. We also used an additional blind test set of 87 mutations across five homology models as proposed previously (Pires and Ascher, 2016b). The datasets used to train and validate mCSM-AB2 are available on the mCSM-AB2 web server.
2.2 Feature engineering
Three main classes of features were used in mCSM-AB2 as evidence to train and test predictive models via supervised learning—structural, evolutionary and energy-based terms. Graph-based signature are calculated to model the wild-type residue environment.
These represent distance patterns between different atom types as cumulative distributions of distances, which we previously show encode both its physicochemical aspects and geometry (Pires et al., 2014a, 2016a; Pires and Ascher, 2016a, 2017; Rodrigues et al., 2018a). In order to calculate structure-based features for mutants, we implemented BuildModel of FoldX for high quality models. Additionally, the changes in pharmacophores due to the mutation are also modelled as a feature vector. These pharmacophore changes calculated the difference in atom counts per class (hydrophobic, positive charge, negative charge, hydrogen acceptor, hydrogen donor, aromatic, sulphur and neutral) between wild-type and mutant residues. Additional structural information was also taken into account, including the change in molecular interactions upon mutation as calculated by Arpeggio (Jubb et al., 2017), the distance change of the mutation to the Ab–antigen interface, and the change of relative solvent accessible (RSA) area upon mutation using DSSP (Touw et al., 2015). Evolutionary-based information was integrated by calculating the difference of evolutionary scores between wild-type and mutant using PAM30-based position-specific scoring matrices (PSSM) (Altschul et al., 1997). An energy-based term was also generated using FoldX (Stricher et al., 2005) force fields to calculate the difference upon mutation in potential energy between the wild-type and mutant structures, expressed in kcal/mol.
2.3 Machine learning methods
Using the collected experimental data describing the effects of missense mutations on Ab–antigen affinity and calculated features, different supervised learning algorithms available on the Scikit-learn library for Python (Pedregosa et al., 2011) were evaluated, including Extra Trees (Geurts et al., 2006), Random Forest (Breiman, 2001), Gradient Boost (Friedman, 2002) and XGBoost (Chen and Guestrin, 2016) regression. Predictive models were trained using five times stratified 10-fold cross-validation to avoid sampling bias, followed by a blind test. A leave-one-complex out cross-validation procedure was also implemented to assess performance variations for different Ab–antigen complexes. The final model showed comparable performances across the different training schemes including 5-fold, 10-fold, leave-one-complex-out and Jackknife (Wager et al., 2014) validation, as shown in Supplementary Table S2.
2.4 Evaluation metrics
The performance of individual models was assessed using the Pearson’s correlation coefficient and root mean square error (RMSE), considering performances on both cross-validation and blind tests. The performance of the model was also assessed on 90% of the data after removing 10% of worst predicted cases to evaluate effects of outliers on model accuracy.
3 Results
In order to evaluate the performance of mCSM-AB2, we devised a series of experiments. The first aim was to assess the contribution of individual feature components to predictive performance as well as their combination. mCSM-AB2 was further tested on blind tests and its performance was compared with available methods.
3.1 Quantitative assessment of Ab–antigen affinity changes upon mutation
Building upon the previous version of mCSM-AB, we have integrated new structure-based features, energy-based terms and evolutionary scores with our graph-based signatures to better model the changes of topological and physicochemical properties on Ab–antigen affinity induced by missense mutations. Supplementary Table S2 shows the predictive performance of the individual feature classes, given as Pearson’s correlation coefficient, for different validation procedures, including 5- and 10-fold cross-validation, as well as Jackknife validation.
The best performing individual class of features was the graph-based signatures, contributing to a correlation of ρ = 0.65 (RMSE of 2.14 kcal/mol) on 10-fold cross-validation, followed by the difference in contacts made by wild-type and mutant residues, which achieved a correlation of ρ = 0.60 (RMSE of 2.40 kcal/mol), highlighting the important role of inter-residue interactions on driving Ab–antigen affinity and recognition. Pharmacophore modelling was also an important feature class, achieving a correlation of ρ = 0.50 (RMSE of 3.12 kcal/mol). Complementary structure-based information was also integrated to the method, even with modest performance, including the change of the RSA upon mutation (ρ = 0.16 and RMSE of 10.92 kcal/mol), the change of distance from mutation site to the antigen interface (ρ = 0.26 and RMSE of 6.64 kcal/mol).
Other two features incorporated on this new and updated version of the method were energy potential terms calculated using FoldX and sequence-based evolutionary information encoded in PSSM scoring matrices. These features contributed individually to a predictive performance of ρ = 0.26 (RMSE of 6.61 kcal/mol) and ρ = 0.42 (RMSE of 3.95 kcal/mol), respectively.
It is interesting to notice that there seems to be little correlation between the different classes of selected features, as shown in Supplementary Figure S2, especially to the new evolutionary and energy-based attributes, indicating they were likely contributing to the predictive model with non-redundant, novel information. In addition, regardless of lower performance of evolutionary- and energy-based features, those have greater importance on the mCSM-AB2 model which indicates those two features high chance to give synergistic effect with other features, not by themselves (Supplementary Figure S3).
By combining the different feature classes to train a regressor algorithm/model, we obtained an improved and optimized model capable of accurately and quantitatively predicting effects of mutations on Ab–antigen binding affinity across eight different algorithms, achieving a Pearson’s correlation coefficient of ρ = 0.73 (RSME of 1.68 kcal/mol) from Extra Tress algorithm (Supplementary Fig. 2A, Table S3) on 10-fold cross-validation. This model was significantly different (P ≪ 0.05 by Diebold–Mariano test) compared with the null hypothesis using the average of all values as the prediction (RMSE = 1.80 kcal/mol), the average of just the experimentally measured changes in binding affinity (RMSE = 2.07 kcal/mol), and by randomly scrambling the ΔΔG 10 times to keep the same data distribution (RMSE = 2.56 kcal/mol). The performance of the method increases to ρ = 0.84 on 90% of the data and was not significantly different when either 5-fold cross-validation or Jackknife validation were used, providing additional confidence in the model.
Compared with earlier mCSM-AB, we implemented additional features from both wild-type and mutant structures which demand more computational cost, but those features improved the performance on training and two blind tests (Supplementary Table S4). The reliability of the model structures obtained through FoldX was assessed by comparing with seven experimental mutant structures (Supplementary Table S5). The modelled structures used in mCSM-AB2 showed a low average Cα RMSD of 0.13 Å.
3.2 Comparative performance and blind tests
In order to put mCSM-AB2 prediction results into context, we have carried out a performance comparison with other available methods using a non-redundant blind test composed of 754 mutations with experimentally measured changes in binding affinity. mCSM-AB2 significantly outperformed alternative approaches, achieving a Pearson’s correlation coefficient of ρ = 0.64 (P ≤ 0.0001, as depicted in Table 1 and Fig. 2B), showing that not only it was able to accurately predict Ab–antigen binding affinity changes but also presented a significant improvement in comparison with its previous version (ρ = 0.42). This performance was comparable to the cross-validation performance, increasing our confidence in the method’s generalization capabilities.
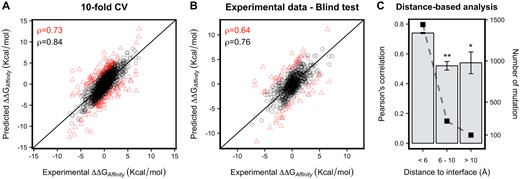
Performance of mCSM-AB2 in predicting Ab–antigen affinity changes upon mutation. mCSM-AB2 achieved a Pearson’s correlation of ρ = 0.73 (RMSE = 1.68) on 10-fold cross-validation (A), ρ = 0.64 (RMSE = 1.85) on a non-redundant experimental dataset for a model trained on the AB-BIND dataset (B). Performance of mCSM-AB2 after excluding the 10% largest errors (red triangles) are shown as black circles. (C) Through 10 times of 10-fold cross-validation runs, mCSM-AB2 achieved a Pearson’s correlation of 0.74 (σ = 0.004), 0.52 (σ = 0.029) and 0.54 (σ = 0.073) on mutations whose distances to their Ab–antigen binding interfaces are less than 6 Å (1442 mutations), between 6 and 10 Å (269 mutations) and greater than 10 Å (99 mutations), respectively. Fisher’s r-to-z transformation was used to compare Pearson’s correlations from different size of mutation (* P ≤ 0.001 and ** P ≤ 0.0001). (Color version of this figure is available at Bioinformatics online.)
Performance comparison between mCSM-AB2 and available methods
| Method | Pearson’s correlation | ||
|---|---|---|---|
| Training | Blind test | ||
| AB-BIND | Experimental set | Homology model | |
| bASA | 0.22a,*** | 0.29*** | 0.41*** |
| dDFIRE | 0.19a,*** | 0.31*** | 0.53*** |
| DFIRE | 0.31a,*** | 0.38*** | 0.52** |
| FoldX | 0.34a,*** | 0.26*** | 0.45*** |
| Discovery Studio | 0.45a,*** | 0.31*** | 0.53** |
| mCSM-PPI | 0.35a,*** | 0.32*** | 0.26*** |
| mCSM-AB | 0.56a,*** | 0.42*** | 0.54* |
| mCSM-AB2 | 0.76 | 0.64 | 0.77 |
| Method | Pearson’s correlation | ||
|---|---|---|---|
| Training | Blind test | ||
| AB-BIND | Experimental set | Homology model | |
| bASA | 0.22a,*** | 0.29*** | 0.41*** |
| dDFIRE | 0.19a,*** | 0.31*** | 0.53*** |
| DFIRE | 0.31a,*** | 0.38*** | 0.52** |
| FoldX | 0.34a,*** | 0.26*** | 0.45*** |
| Discovery Studio | 0.45a,*** | 0.31*** | 0.53** |
| mCSM-PPI | 0.35a,*** | 0.32*** | 0.26*** |
| mCSM-AB | 0.56a,*** | 0.42*** | 0.54* |
| mCSM-AB2 | 0.76 | 0.64 | 0.77 |
Note: Pearson’s correlation coefficient of each of the methods were compared with mCSM-AB2 by Fisher’s r-to-z transformation (*P ≤ 0.05, **P ≤ 0.001 and ***P ≤ 0.0001).
From Sirin et al. (2016).
Performance comparison between mCSM-AB2 and available methods
| Method | Pearson’s correlation | ||
|---|---|---|---|
| Training | Blind test | ||
| AB-BIND | Experimental set | Homology model | |
| bASA | 0.22a,*** | 0.29*** | 0.41*** |
| dDFIRE | 0.19a,*** | 0.31*** | 0.53*** |
| DFIRE | 0.31a,*** | 0.38*** | 0.52** |
| FoldX | 0.34a,*** | 0.26*** | 0.45*** |
| Discovery Studio | 0.45a,*** | 0.31*** | 0.53** |
| mCSM-PPI | 0.35a,*** | 0.32*** | 0.26*** |
| mCSM-AB | 0.56a,*** | 0.42*** | 0.54* |
| mCSM-AB2 | 0.76 | 0.64 | 0.77 |
| Method | Pearson’s correlation | ||
|---|---|---|---|
| Training | Blind test | ||
| AB-BIND | Experimental set | Homology model | |
| bASA | 0.22a,*** | 0.29*** | 0.41*** |
| dDFIRE | 0.19a,*** | 0.31*** | 0.53*** |
| DFIRE | 0.31a,*** | 0.38*** | 0.52** |
| FoldX | 0.34a,*** | 0.26*** | 0.45*** |
| Discovery Studio | 0.45a,*** | 0.31*** | 0.53** |
| mCSM-PPI | 0.35a,*** | 0.32*** | 0.26*** |
| mCSM-AB | 0.56a,*** | 0.42*** | 0.54* |
| mCSM-AB2 | 0.76 | 0.64 | 0.77 |
Note: Pearson’s correlation coefficient of each of the methods were compared with mCSM-AB2 by Fisher’s r-to-z transformation (*P ≤ 0.05, **P ≤ 0.001 and ***P ≤ 0.0001).
From Sirin et al. (2016).
Comparison of mCSM-AB2 performance across the training set also showed it performed significantly better than other methods that have been used to guide rational Ab engineering (Table 1). Interestingly, there were only weak correlations between mCSM-AB2 and other Ab engineering methods (Supplementary Fig. S4), including the original method, highlighting its use of complementary but distinguishing information, and suggesting that a consensus predictor might be informative.
The experimental datasets were enriched in mutations located at the antigen interface (>80% within 6 Å as shown in Fig. 2C), which is not surprising since many experiments have focused on variations in the CDR loops with alanine scanning (>60% of mutations in the dataset are to alanine). The distance from a mutation site to the Ab–antigen interface influenced on the performance of mCSM-AB2. Comparing performance on mutations less than 6 Å, 6–10 Å and greater than 10 Å away from the antigen interface, mCSM-AB2 achieved a Pearson’s correlation of 0.74 (σ = 0.004), 0.52 (σ = 0.029) and 0.54 (σ = 0.073), respectively. This deterioration of the performance on mutations located further away from the interface may be due to the limited number of distal mutations in the training set. As a result of the distance-based analysis, the mCSM-AB2 web server gives users a confidence level of prediction, high or moderate, depending on the distance between the mutation and Ab–antigen binding interface.
While mutations to alanine were inherently enriched in the dataset, the performance of mCSM-AB2 was consistent across mutations to any residue (Supplementary Table S6). This can be further supported by the analysis of the experimental blind test results showing mCSM-AB2 outperforms all other methods across all types of mutations (Supplementary Fig. S5).
An earlier study (Sinha et al., 2002) suggested several experimental ΔΔGs from the HyHEL-10 Fab and lysozyme complex (PDB: 3HFM), which were measured by indirect methods such as spectroscopic inhibition assay (IASP) and spectroscopic method (SP), presented a large discrepancy with ΔΔG from direct method such as surface plasmon resonance (SPR). In order to measure the contribution of each of Ab–antigen complexes on the performance of mCSM-AB2, we conducted the leave-one-complex-out cross-validation on the 60 Ab–antigen complexes. Notably, the mutations from 3HFM presented a large portion of outliers in both 10-fold and leave-one-complex-out cross validations showing 31 and 17 out of 181 worst predicted data points, respectively (Supplementary Table S7). The overall performance on leave-one-complex-out (Pearson’s correlation of ρ = 0.70), however, was comparable with the 10-fold cross-validation results (Pearson’'s correlation of ρ = 0.73), further demonstrating the robustness of the method.
3.3 Performance on homology models
As experimental crystal structures might not always be available, we also wanted to compare the performance of mCSM-AB2 on predicting effects of mutations on Ab–antigen binding affinity using homology models. We used a previously proposed homology model dataset (Sirin et al., 2016) of 87 experimentally measured changes in binding affinity upon mutation across five homology models of the corresponding Ab–antigen complex. The mCSM-AB2 predictions correlated well with the experimental values (ρ = 0.77, RMSE = 1.66), and was significantly more accurate than all other predictive methods analyzed (Table 1). This highlights the versatility and robustness of the mCSM-AB2 predictions, and its applicability even in the absence of an experimental structure of an Ab–antigen complex.
3.4 mCSM-AB2 web server
We have developed a web server to provide the functionalities of mCSM-AB2 in an intuitive way, increasing reproducibility and facilitating large-scale analyses. The front-end was designed with Bootstrap framework version 4.1 and the back-end was based on Python 2.7 via the Flask framework version 1.0.2 on a Linux server running Apache. It allows users to upload Ab–antigen complexes (in PDB format) and either analyze specific mutations provided by the user, or systematically evaluate mutations across the entire Ab–antigen interface via either alanine scanning or saturation mutagenesis, facilitating, for instance, the identification of mutations that are more likely to increase affinity, aiding the rational design of Abs. The results pages allow easy visualization of the alanine-scanning and saturation-mutagenesis predictions mapped to the 3D structure as well as heat-mapped tables (Fig. 3). Users are able to check information such as distance to binding interface and Chothia annotation calculated by ANARCI (Dunbar and Deane, 2016) and download all results including the predictions as a CSV file, and the provided PDB files with the predicted changes in binding affinity mapped to the B-factor column.
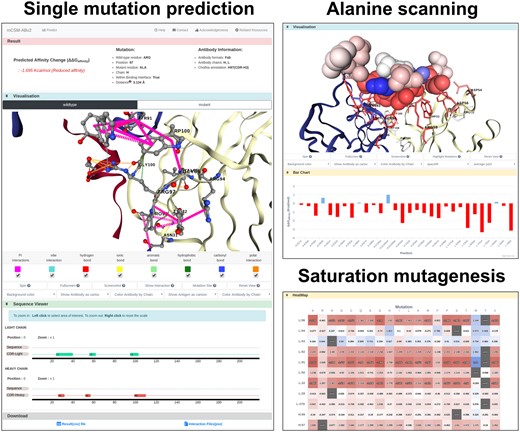
mCSM-AB2 web server result pages. Single mutation prediction (left) provides predicted ΔΔGAffinity and interaction changes upon mutation via a 3D molecular viewer for both wild-type and mutant. Alanine scanning (right top) describes mutational effects on interface residues with molecular viewer and bar charts. In saturation mutagenesis analysis (right bottom), users can check Ab–antigen affinity changes for each of the 19 possible mutations for each interface residues
4 Conclusions
The ability to predict favourable Ab–antigen mutations is a crucial, but non-trivial, challenge to help guide routine affinity maturation. While a number of successful computational-guided Ab development examples have been published in recent years, computational tools haven’t had yet transformative effects for Ab engineering due to limited accuracy of available computational methods.
mCSM-AB2 is a computational approach that leverages both sequence and structural information to allow users to accurately assess the effects of single-point mutations on Ab–antigen binding affinity. Across all training and blind test evaluations, mCSM-AB2 significantly outperformed all currently used Ab mutational analysis approaches, using both experimental structures and homology models, highlighting its potential power to help guide Ab development. This also highlights the power of our graph-based signatures in terms of predicting mutational effects on Ab–antigen affinities by efficiently representing structural environment of wild-type and mutant residues, but also show the importance of considering evolutionary aspects, energetic terms and inter-residue interactions to better understand molecular recognition.
We believe that mCSM-AB2 will be a powerful tool to not only streamline Ab development and engineering but also providing better insight into the effects of mutations in Ab–antigen interfaces, including escape mutations. A user-friendly web server implementing mCSM-AB2 functionalities was implemented and is freely available at http://biosig.unimelb.edu.au/mcsm_ab2, facilitating large-scale analysis of entire Ab–antigen interfaces.
Funding
Y.M. and C.H.M.R were funded by the Melbourne Research Scholarship. D.B.A and D.E.V.P were funded by a Newton Fund RCUK-CONFAP Grant awarded by The Medical Research Council (MRC) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) [MR/M026302/1]; Conselho Nacional de Desenvolvimento Cientı´fico e Tecnológico (CNPq); the Jack Brockhoff Foundation [JBF 4186, 2016]; and a C. J. Martin Research Fellowship from the National Health and Medical Research Council (NHMRC) of Australia [APP1072476]. Supported in part by the Victorian Government’s OIS Program.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}