





Abstract
Shotgun DNA sequencing provides sensitive detection of all 182 HPV types in tissue and body fluid. However, existing computational methods either produce false positives misidentifying HPV types due to shared sequences among HPV, human and prokaryotes, or produce false negative since they identify HPV by assembled contigs requiring large abundant of HPV reads.
We designed HPViewer with two custom HPV reference databases masking simple repeats and homology sequences respectively and one homology distance matrix to hybridize these two databases. It directly identified HPV from short DNA reads rather than assembled contigs. Using 100 100 simulated samples, we revealed that HPViewer was robust for samples containing either high or low number of HPV reads. Using 12 shotgun sequencing samples from respiratory papillomatosis, HPViewer was equal to VirusTAP, and Vipie and better than HPVDetector with the respect to specificity and was the most sensitive method in the detection of HPV types 6 and 11. We demonstrated that contigs-based approaches had disadvantages of detection of HPV. In 1573 sets of metagenomic data from 18 human body sites, HPViewer identified 104 types of HPV in a body-site associated pattern and 89 types of HPV co-occurring in one sample with other types of HPV. We demonstrated HPViewer was sensitive and specific for HPV detection in metagenomic data.
HPViewer can be accessed at https://github.com/yuhanH/HPViewer/.
Supplementary data are available at Bioinformatics online.
1 Introduction
Human papillomavirus (HPV) is a type of double-stranded small DNA virus that causes nearly 610 000 cases of cancers annually in the world (Forman et al., 2012). Many studies have demonstrated that HPV is a vital cause of cervical cancers (Bosch et al., 2002; Ho et al., 1995; Walboomers et al., 1999) and these studies have classified HPV types as high risk and low risk. Munoz et al., grouped HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, 82 as high risk; and HPV 6, 11, 40, 42, 43, 44, 54, 61, 70, 72, 81, 89 were considered as low risk (Muñoz et al., 2003). Because of the variation of HPV prevalence and co-occurrence among different body sites (Ma et al., 2014), other types of HPV not belong to cervical high and low risk types also have potentiality to cause diseases in other body sites. HPV has been linked to other cancers including cancers of the oropharynx (Gillison, 2008), head, neck (Gillison et al., 2000; Parfenov et al., 2014; Tang et al., 2013). Of particular concern, the incidence of HPV-associated oropharynx cancer (Chaturvedi et al., 2011) is growing very rapidly. Furthermore, HPV DNA has been detected in cancers of the lung, colon, esophagus and urinary bladder (Cheng et al., 2001; Furihata et al., 1993; Kawaguchi et al., 2000; Perez et al., 2005). The range of HPV types increases beyond the high and low risk HPV in the uterine cervix when others body sites were included in the consideration. Currently, 210 types of HPV have been identified in the International HPV Reference Center (http://www.hpvcenter.se) and this number is increasing monthly. There are 182 types of HPV with complete genomes sequences in the PapillomaVirus Episteme (PaVE) (https://pave.niaid.nih.gov/).
The traditional clinical HPV detection methods can be classified into three groups: nucleic acid-hybridization assays, nucleic-acid amplification and antibody-based assays (Abreu et al., 2012). Nucleic acid-hybridization assays make use of in situ hybridization, which can detect the 13 most high-risk HPV genotypes, including types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59 and 68 through a biotinylated-probe cocktail (GenPoint HPV Probe Cocktail, Dako) or other HPV types with custom designed probes (Ang et al., 2010). Inno-LiPA (van Hamont et al., 2006) can detect 32 types of HPV by PCR amplification of a 65 bp region of the conserved L1 gene and then performing reverse line blot hybridization to identify specific HPV types. A real-time TaqMan PCR assay can also be used for HPV detection through determining the presence of mRNA of E6 genes of HPV (Koshiol et al., 2011). PapilloCheck, developed by Greiner Bio-One, is a nucleic-acid amplification method, which amplifies the E1 gene of HPV and can identify 24 types of HPV (Dalstein, Merlin et al. 2009). There are two FDA-approved HPV assays using nucleic-acid amplification. Cobas® HPV Test by Roche (Indianapolis, IN, USA) can detect 14 types of high-risk HPV DNA through PCR and fluorescence (Stoler et al., 2011). Aptima® by GenProbe (Woburn, MA, USA) targets high-risk HPV mRNA from E6/E7 genes by transcription-mediated amplification (Dockter et al., 2009). An indirect assay for HPV16 infection is available by immunohistochemistry of expression of a human gene, p16, because there is an overexpression of p16 resulting from HPV-16 integration into the host genome and disruption of the retinoblastoma pathway (Begum et al., 2003). More recently, Lavezzo et al. (2016) proposed a new HPV genotyping method depending on conserved PCR primers for the E6/E7 region (Lavezzo et al., 2016), but this new method is limited to detection of only high-risk types of HPV.
These methods, although covering mainly the 26 high/low risk HPV types, are sufficient to detect all HPV types related to cervical cancer (Shah et al., 2016). Our understanding of the causality of HPV in other cancers is mainly derived from surveys by using the cervical HPV detection methods. However, HPV type distribution in other body sites differs greatly from the uterine cervix (Ma et al., 2014) and there have been no methods or kits specially designed for detecting HPV types found at these body sites. Considering HPV prevalence in cancers outside of the uterine cervix could be underestimated due to the inabilities of cervical HPV kit to detect all HPV types, so a broad range method to detect all HPV types is needed to allow a complete evaluation of the role of HPV in cancers outside of the uterine cervix.
Shotgun sequencing of human tissue samples or body fluids is a robust tool which can broaden the narrow spectrum of the traditional HPV detection approaches. It depends on bioinformatics pipelines to identify and genotype HPV reads from a large pool of human and microbial DNA sequences. Johannsson et al. (2013) applied MEGABLAST to filter out human and bacteria reads and performed de novo assembly to obtain long contigs and used BLASTn against GenBank to identify HPV (Johansson et al., 2013). Ma et al. (2014) applied a HPV genotyping framework through BLAST to a local reference HPV database for detection of HPV reads in datasets generated from a variety of human body sites by whole genome shotgun sequencing (WGS) (Ma et al., 2014). BLAST is a powerful but time-consuming tool (Altschul et al., 1990) and it is very inefficient for processing millions of short DNA fragments from metagenomic data. HPVDetector, developed in 2015 (Chandrani et al., 2015), depends on the Burrows-Wheeler Aligner (Li and Durbin, 2009) to match shotgun reads to their reference genome database. There are also several software programs designed for identifying all viruses including HPV in WGS data, such as Metavir2 (Roux et al., 2014), VirSorter (Roux et al., 2015), VirusTAP (Yamashita et al., 2016), VirusScan (Cao et al., 2016), Vipie (Lin et al., 2017), VIP (Li et al., 2016) and VirFinder (Ren et al., 2017). Table 1 provides a summary of important characteristics of 9 different programs available.
Comparison of current HPV or virome detection tools
| Detection tool | Function | Web based | Input data | Methods of virus identification | De novo assembly | Database |
|---|---|---|---|---|---|---|
| HPViewer | Genotyping and quantification of HPV with non-specific repeats masked. | No | Raw reads | Bowtie2 | No | Repeat-masked and homology-masked HPV genomes |
| HPVDetector | Detecting of HPV and identifying chromosomal integration sites | No | Raw reads | BWA | No | Multiple HPV genomes and human genome |
| VirusTAP | Identification of viral genome sequences after subtraction of host and bacteria-related reads | Yes | Raw reads | BLAST | Yes | Customized viral nucleotide/protein sequences from the NCBI nt/nr database excluding bacteriophages, human genome, ribosomal RNAs, bacterial genome sequences, the latest host organisms genome sequences |
| Vipie | Parallel analysis of multiple metagenomic samples for viruses identification | Yes | Raw reads | BLAST | Yes | A custom database with 20 759 viruses, human genome, ribosomal DNA of bacteria, archaea and fungi |
| VirusScan | Investigation of the viral presence in human tumors | No | Raw reads | BWA | No | A custom virus database containing clustered viral sequences from NCBI NT database, human genome |
| VIP | One-touch pipeline for metagenomic virus identification | No | Raw reads | Bowtie2 | Yes | Virus nucleotide from ViPR, IRD, Refseq viral, DDBJ, EMBL and GenBank, viral protein databases from Refseq, human genome, bacterial genome from GOTTCHA |
| Metavir2 | Viral detection of metagenomics | Yes | Raw reads or contigs | BLAST, HMM | Either | Virus DNA and protein database from RefSeq NCBI taxonomy, kmer frequency pattern, PFAM protein domain database |
| VirSorter | Metagenomic virus identification in both reference dependent and independent manners | No | Contigs | BLAST, HMM | No | Virus proteins from RefSeq, viral sequences sampled from freshwater, seawater and human gut, lung and saliva, PFAM protein domain database |
| VirFinder | Identification of viral sequences by k-mer analysis | No | Contigs | k-mer frequency based machine learning | No | Virus k-mer signatures |
| Detection tool | Function | Web based | Input data | Methods of virus identification | De novo assembly | Database |
|---|---|---|---|---|---|---|
| HPViewer | Genotyping and quantification of HPV with non-specific repeats masked. | No | Raw reads | Bowtie2 | No | Repeat-masked and homology-masked HPV genomes |
| HPVDetector | Detecting of HPV and identifying chromosomal integration sites | No | Raw reads | BWA | No | Multiple HPV genomes and human genome |
| VirusTAP | Identification of viral genome sequences after subtraction of host and bacteria-related reads | Yes | Raw reads | BLAST | Yes | Customized viral nucleotide/protein sequences from the NCBI nt/nr database excluding bacteriophages, human genome, ribosomal RNAs, bacterial genome sequences, the latest host organisms genome sequences |
| Vipie | Parallel analysis of multiple metagenomic samples for viruses identification | Yes | Raw reads | BLAST | Yes | A custom database with 20 759 viruses, human genome, ribosomal DNA of bacteria, archaea and fungi |
| VirusScan | Investigation of the viral presence in human tumors | No | Raw reads | BWA | No | A custom virus database containing clustered viral sequences from NCBI NT database, human genome |
| VIP | One-touch pipeline for metagenomic virus identification | No | Raw reads | Bowtie2 | Yes | Virus nucleotide from ViPR, IRD, Refseq viral, DDBJ, EMBL and GenBank, viral protein databases from Refseq, human genome, bacterial genome from GOTTCHA |
| Metavir2 | Viral detection of metagenomics | Yes | Raw reads or contigs | BLAST, HMM | Either | Virus DNA and protein database from RefSeq NCBI taxonomy, kmer frequency pattern, PFAM protein domain database |
| VirSorter | Metagenomic virus identification in both reference dependent and independent manners | No | Contigs | BLAST, HMM | No | Virus proteins from RefSeq, viral sequences sampled from freshwater, seawater and human gut, lung and saliva, PFAM protein domain database |
| VirFinder | Identification of viral sequences by k-mer analysis | No | Contigs | k-mer frequency based machine learning | No | Virus k-mer signatures |
Comparison of current HPV or virome detection tools
| Detection tool | Function | Web based | Input data | Methods of virus identification | De novo assembly | Database |
|---|---|---|---|---|---|---|
| HPViewer | Genotyping and quantification of HPV with non-specific repeats masked. | No | Raw reads | Bowtie2 | No | Repeat-masked and homology-masked HPV genomes |
| HPVDetector | Detecting of HPV and identifying chromosomal integration sites | No | Raw reads | BWA | No | Multiple HPV genomes and human genome |
| VirusTAP | Identification of viral genome sequences after subtraction of host and bacteria-related reads | Yes | Raw reads | BLAST | Yes | Customized viral nucleotide/protein sequences from the NCBI nt/nr database excluding bacteriophages, human genome, ribosomal RNAs, bacterial genome sequences, the latest host organisms genome sequences |
| Vipie | Parallel analysis of multiple metagenomic samples for viruses identification | Yes | Raw reads | BLAST | Yes | A custom database with 20 759 viruses, human genome, ribosomal DNA of bacteria, archaea and fungi |
| VirusScan | Investigation of the viral presence in human tumors | No | Raw reads | BWA | No | A custom virus database containing clustered viral sequences from NCBI NT database, human genome |
| VIP | One-touch pipeline for metagenomic virus identification | No | Raw reads | Bowtie2 | Yes | Virus nucleotide from ViPR, IRD, Refseq viral, DDBJ, EMBL and GenBank, viral protein databases from Refseq, human genome, bacterial genome from GOTTCHA |
| Metavir2 | Viral detection of metagenomics | Yes | Raw reads or contigs | BLAST, HMM | Either | Virus DNA and protein database from RefSeq NCBI taxonomy, kmer frequency pattern, PFAM protein domain database |
| VirSorter | Metagenomic virus identification in both reference dependent and independent manners | No | Contigs | BLAST, HMM | No | Virus proteins from RefSeq, viral sequences sampled from freshwater, seawater and human gut, lung and saliva, PFAM protein domain database |
| VirFinder | Identification of viral sequences by k-mer analysis | No | Contigs | k-mer frequency based machine learning | No | Virus k-mer signatures |
| Detection tool | Function | Web based | Input data | Methods of virus identification | De novo assembly | Database |
|---|---|---|---|---|---|---|
| HPViewer | Genotyping and quantification of HPV with non-specific repeats masked. | No | Raw reads | Bowtie2 | No | Repeat-masked and homology-masked HPV genomes |
| HPVDetector | Detecting of HPV and identifying chromosomal integration sites | No | Raw reads | BWA | No | Multiple HPV genomes and human genome |
| VirusTAP | Identification of viral genome sequences after subtraction of host and bacteria-related reads | Yes | Raw reads | BLAST | Yes | Customized viral nucleotide/protein sequences from the NCBI nt/nr database excluding bacteriophages, human genome, ribosomal RNAs, bacterial genome sequences, the latest host organisms genome sequences |
| Vipie | Parallel analysis of multiple metagenomic samples for viruses identification | Yes | Raw reads | BLAST | Yes | A custom database with 20 759 viruses, human genome, ribosomal DNA of bacteria, archaea and fungi |
| VirusScan | Investigation of the viral presence in human tumors | No | Raw reads | BWA | No | A custom virus database containing clustered viral sequences from NCBI NT database, human genome |
| VIP | One-touch pipeline for metagenomic virus identification | No | Raw reads | Bowtie2 | Yes | Virus nucleotide from ViPR, IRD, Refseq viral, DDBJ, EMBL and GenBank, viral protein databases from Refseq, human genome, bacterial genome from GOTTCHA |
| Metavir2 | Viral detection of metagenomics | Yes | Raw reads or contigs | BLAST, HMM | Either | Virus DNA and protein database from RefSeq NCBI taxonomy, kmer frequency pattern, PFAM protein domain database |
| VirSorter | Metagenomic virus identification in both reference dependent and independent manners | No | Contigs | BLAST, HMM | No | Virus proteins from RefSeq, viral sequences sampled from freshwater, seawater and human gut, lung and saliva, PFAM protein domain database |
| VirFinder | Identification of viral sequences by k-mer analysis | No | Contigs | k-mer frequency based machine learning | No | Virus k-mer signatures |
One consideration for identifying HPV with short reads in WGS data is false positivity caused by homologous sequences and/or repeats shared among the host, microbes and HPV. In addition, genotyping can be inaccurate when HPV reads detected are shared by more than one HPV type. One approach to reduce false positivity is using de novo assembly to generate large contigs that cover a larger region of HPV genome beyond the shared region. Of the nine programs, six applied or required the de novo assembly approach, including VirSorter, VirFinder, Metavir2, VirusTAP, Vipie and VIP. However, contigs from de novo assembly can be constructed only if the data have sufficient coverage, limiting its capability of detecting HPV in samples in which HPV reads are too few to form a contig. Another approach to reduce false positivity is to filter out host and bacterial genome sequences. For example, VIP, VirusTAP and VirusScan subtract the input DNA fragments which can align to the host genome before searching for HPV DNA. This strategy has two shortcomings because of the large size of host genome. It not only takes long time to align input DNA fragments to the host genome but also needs large storage space for the host genome database for local use. In addition, this approach does not reduce genotyping errors due to homology among closely related HPV genotypes. HPVDetector, the program specially designed for HPV detection, does not consider the false positive issues from the host genome and homology among different HPV types.
In the present study, we developed a new HPV detection program—HPViewer that reduces false detection of HPV DNA by masking simple repeats commonly shared among the human, prokaryotes genomes and homologous sequences shared by different HPV types. We evaluated the sensitivity and specificity of HPViewer using 100 100 simulation samples, and in a WGS dataset from patients with recurrent respiratory papillomatosis which are known to be associated with HPV6/11 (Gissmann et al., 1983), compared the performance of HPViewer with HPVDetector, VirusTAP and Vipie. We also applied HPViewer to define HPV prevalence distribution and explore the HPV co-occurrence patterns in different body sites of healthy samples from the Human Microbiome Project (HMP).
2 Materials and methods
2.1 Two HPV genome databases in HPViewer
We downloaded all 182 HPV reference genomes from PaVE for this study. Bowtie2 (version 2.2.7) is the alignment tool utilized in this study (Langmead and Salzberg, 2012). All metagenomic reads were aligned to our customized HPV databases through bowtie2 in the end-to-end, sensitive mode.
We created two local HPV databases with two different masking strategies, repeat-mask and homology-mask. For the repeat-mask database, we used RepeatMasker to replace the low complexity and simple repeats regions of all HPV genomes with ‘N’. For the homology-mask database which was inspired by Metaphlan (Segata et al., 2012), we created a type-specific HPV database by masking homologous sequences shared among different HPV types, and then further masked the repeats using Repeat Masker (Supplementary Fig. S1). There were three steps for the construction of homology-mask database. First, all 100 bp DNA fragments from each complete HPV genome generated by EMBOSS (Rice et al., 2000), were aligned to all other types of HPV with a 90% identity threshold by bowtie2 (bowtie2 parameters: -a –score-min L, 0.6, 0.6). Then we masked the matching regions on the genomes (Supplementary Fig. S1). Finally, after all homologous regions were masked, RepeatMasker was also applied for all processed HPV genomes to mask low complexity and simple repeats regions (Supplementary Fig. S1). For repeat-mask and homology-mask databases, the length of HPV genomes was not changed and only some fragments were replaced as ‘N’, and we called non-N sequences of HPV genome as the effective genome. The distribution of effective genome size of original HPV, repeat-mask and homology-mask was generated by the R package, ggplot2 (Wickham, 2009).
We validated the repeat-mask database by BLASTn against genomes of human (GCRh38) and prokaryotes (Prokaryotic RefS eq 112 reference gnomes and 1669 representative genomes) (https://www.ncbi.nlm.nih.gov/genome/browse/reference/), and no matches were found with identity > 90% over an alignment region > 50 bp. The circos plot of shared sequences between HPV and human, prokaryotes was generated by Circos 0.69 (Krzywinski et al., 2009).
2.2 Construction of the homology tree among 182 types of HPV and the hybrid-mask of HPViewer
In order to explore the sequence similarity among HPV types, we selected from each genome only the 100 bp genome fragments with >90% identity to two or more HPV types from each HPV type, all other bases in the genomes were masked as N (Supplementary Fig. S1). There were 29 types of HPV without any 100 bp regions that matched other types. The selected portions from the remaining 153 HPV genomes were multiple-aligned with MUSCLE 3.8.31 (Edgar, 2004). The pairwise distance matrix was calculated by MEGA7 (Kumar et al., 2016) and the maximum likelihood tree was built with RAxML 8.2.9 under a GTRCAT substitution model with 1000 bootstrapping replicates (Stamatakis, 2006). The homology tree with a midpoint root was visualized by FigTree v1.4.3 (Rambaut, 2012) (Supplementary Fig. S2).
For the hybrid-mode of HPViewer, first, the repeat-mask mode is used to identify all HPV types in a sample. We set the threshold of detection of one HPV type in a sample as two different aligned reads covering at least 150 bp of a single HPV type reference genome, (Supplementary Fig. S4). We used SAMtools depth (Li et al., 2009) to obtain the coverage for each position of mapped HPV genomes. When the length of the covered positions of the mapped reads on a single HPV type is smaller than 150, we discard that HPV type as false positive. When the covered length is above 150 bp, we considered it as detected.
When only a single HPV type is detected in a sample, there is no chance for false positives from other HPV types, so it is considered as a true positive. When multiple types of HPV are detected in a sample, the HPV types are checked if they are close to each other (the homology distance < 0.35) using the pair-wise homology distance matrix. Distantly related HPV types are reported directly in the HPV profile. The closely related HPV types are required to be re-tested, thus HPV reads generated from repeat-mode output bam file by BEDtools (Quinlan and Hall, 2010) are re-aligned to the homology-mask database. Only similar HPV types detected by homology-mask mode are also added into the HPV profile.
2.3 Simulation of HPV shotgun sequencing data with Grinder
2.4 Detection and genotyping of HPV in patients with recurrent laryngeal papillomatosis using HPViewer
Following approval from the Institutional Review Board at the New York University School of Medicine (study number S13-00119), six patients with pathology-confirmed recurrent respiratory papillomatosis were identified from large pool of patients participating in a large-scale, longitudinal study. Tumor tissue was endoscopically removed, fixed in formalin and embedded in paraffin. The diagnosis of recurrent respiratory papillomatosis was made by histopathological examination of the tumor tissue. To extract DNA, the paraffin-embedded tissue was cut into 20 micron-thick sections. Total genomic DNA was extracted from the unstained tissue sections using BiOstic FFPE Tissue DNA Isolation Kit (Mo Bio Carlsbad, CA).
Oral rinse samples were collected from the same six patients according to the National Health And Nutrition Examination Survey (NHANES) protocol (Born et al., 2014). Briefly, subjects were instructed to swish 5 mL of Scope® mouthwash without gargling for one minute. The oral wash samples were then sealed and stored for no more than one week at 4°C prior to DNA extraction. For DNA extraction, the samples were spun for 10 min at 3200g. DNA in the cell-free supernatant was precipitated with the isopropanol/glycogen solution and pelleted for 10 min at 2000g, as previously described (Born et al., 2014). The pellet was resuspended with 200 µL DNA Hydration Solution (Qiagen).
To evaluate the sensitivity and specificity of HPViewer, we determined the true HPV compositions in these papilloma and oral wash samples. Since only HPV types 6 and 11 have been previously observed in laryngeal papilloma (Gissmann et al., 1983), we conducted standard PCR for HPV6 and 11 using the primers from Tucker et al. (2001) on these 12 samples. We found HPV6 in 4 tumor tissues and 2 matched oral wash samples and HPV11 in 2 tumor tissues. Additional PCR with lower annealing temperature confirmed that HPV6 was present in samples 3W and 7W and that both 3T and 3W were negative for HPV11 (Supplementary Fig. S5).
In the original Bowtie2 screening of these samples on an unmasked HPV database, small numbers of reads matching HPV71 were found in all six oral samples and three tumor samples, as well as HPV19 and 82 in some samples. Inspection of these reads revealed sequences such as TG repeats (Supplementary Fig. S3) which matched to TG repeats in the genome of HPV71. After masking the HPV database with RepeatMasker, no reads matching HPV19, 71, or 82 were found.
HPViewer identified just 2 reads of HPV6 in oral wash samples 3W and 7W. These samples were confirmed as HPV6 positive by PCR. Inspection of the sequence of these reads revealed that they contained the same polymorphisms found in the much larger number of reads in matched tumor samples from the same patients, suggesting a low level of release of HPV from the papilloma into the oral cavity. HPViewer detected just 1 read of HPV11 in sample 3W, but HPV11 was not detected by the PCR in 3W or 3T. Consequently, we have set the detection threshold for HPViewer at 2 different reads per sample for a single HPV type.
2.5 Metagenomic data from Human Microbiome Project
We downloaded 1573 shotgun sequencing metagenomic datasets from Human Microbiome Project (https://hmpdacc.org/hmp/) (Supplementary Table S5). The HMP samples (with human data previously removed) were obtained from 18 body sites, including anterior nares, attached keratinized gingiva, blood, buccal mucosa, ileal pouch, left retroauricular crease, mid vagina, nasopharynx, palatine tonsils, posterior fornix, right retroauricular crease, saliva, stool, subgingival plaque, supragingival plaque, throat, tongue dorsum and vaginal introitus (Supplementary Table S3). The heatmap of HPV prevalence for different body sites were produced by R package, gplot (Warnes et al., 2016). The co-occurrence of HPV for three body sites were generated by Gephi 0.9.1 (Bastian et al., 2009).
3 Results
3.1 HPV genomes share simple repeats with human and prokaryotic genomes
Metagenomic data usually consists of fragments of human and prokaryotic genomes. To detect human and prokaryotic DNA sequences that may interfere with HPV identification, we compared 182 HPV genomes (Van Doorslaer et al., 2016) with human genome (GRCh38) using BLASTn (Altschul et al., 1990) and found 165 118 matches (identity >90%, alignment length >50 bp) between 14 HPV types and all human chromosomes (Fig. 1a, b). All matches were simple repeats and most were TA (83.94%) and TG (15.94%) repeats. Other less abundant repeats, such as TTC, TTCTCC and CATA were also found. In particular, low risk pathogenic HPV types 6, 72, 73 share simple sequence repeats with human chr3, chr1, chr1 and chrY, respectively.
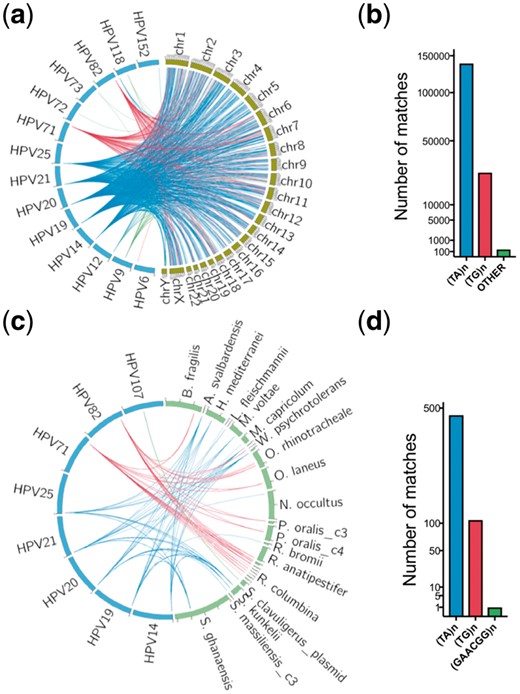
The HPV shared sequences between human and prokaryotic genomes. (a, b) The shared sequences between HPV and the human genome. Each line represents one BLASTn alignment. The blue lines represent TG repeats, and the red lines represent TA repeats and green lines represent other repeats alignments. (c, d) The shared sequences between HPV and prokaryotic genomes. Most were TG and TA repeats. The only other type shared repeat was GAACGG repeat between HPV107 and Streptomyces clavuligerus Plasmid pSCL2 (NZ_CP016560.1). The mapped prokaryotic genomes were listed in Supplementary Table S1
Using the same strategy, we also compared HPV genomes with 1781 prokaryotic genomes (NCBI 112 prokaryotic reference genomes and 1669 NCBI prokaryotic representative genomes) (Supplementary Table S2) and found 575 matches between 8 HPV types and 18 prokaryotic species (Fig. 1c, d), mainly TA (81.22%) and TG (18.61%) repeats plus GAACGG repeats (0.17%). None of the 8 HPV types were high or low risk cervical HPV types. In all 1 375 680 bp of the 182 HPV genomes, simple repeats accounted for 16 359 bp (1.19%).
3.2 Homologous sequences are widely shared among HPV types
Besides homology between HPV and other organisms, homology among HPV types could also interfere with HPV genotyping. To estimate the extent of homology, we aligned each complete HPV genome with genomes of all other type by sliding all possible 100 bp DNA fragments along its entire genome, with a 90% identity threshold. The degree of homology between different types of HPV varied greatly. There are 29 HPV types which lacked homology with any other HPV type, but 85.9% of HPV76 genome was homologous with other HPV types. In the 182 HPV genomes, 368 789 bp (26.81%) were homologous between two or more HPV types.
3.3 Design of HPViewer
We took a novel, masking approach to minimize the impact of the shared sequences on HPV genotyping. Instead of filtering shared sequences by alignment of millions of raw reads in each sample to human and prokaryotic genomes, we masked the simple repeat sequences in the reference HPV genome database with RepeatMasker (Smit, 2015). We then compared these masked HPV genomes with human and prokaryotic genomes and found no matches, indicating that our repeat-mask strategy eliminated false positive calling of human or prokaryotic DNA reads as HPV. Next, we masked all homologous regions shared among HPV types as well as simple repeats as our homology-mask strategy. We found the repeat-mask removed only a few hundreds of nucleotides, while homology-mask considerably changed the distribution of HPV effective genome lengths (Fig. 2). Finally, we built a homology distance matrix and a homology tree only using homologous sequences shared by any other type of HPV (Supplementary Fig. S1).
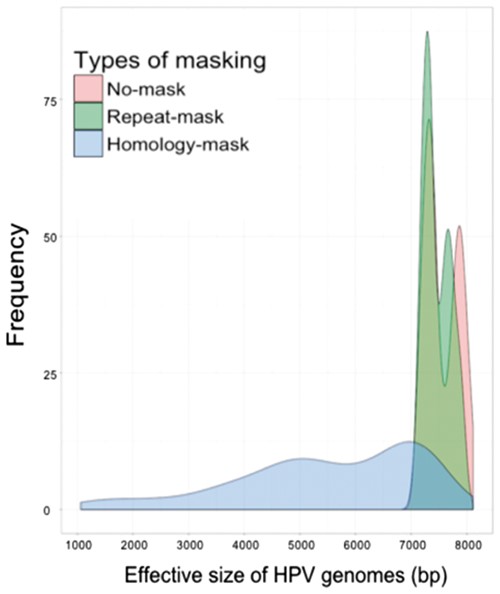
Distribution of HPV effective genome sizes among original HPV genomes, and HPV genomes in repeat-mask, and homology-mask databases. For our mask strategies, the length of HPV genomes was not changed and we called non-N sequences of HPV genome as the effective genome. In sum, the HPV effective genome length ranged from 7100–8104 bp for original genomes, 7100–7995 bp for repeat-mask genomes and 1061–7698 bp for homology-mask genomes
3.4 Estimation of sensitivity and specificity of HPViewer using simulated data
We developed HPViewer for specific detection and quantification of HPV from metagenomic data. Initially, we planned to use a repeat-mask mode to eliminate false positivity caused by human and prokaryotic genomes and a homology-mask mode to prevent errors in genotyping among closely related HPV types.
We evaluated these two modes with 100 100 simulated samples composed of 143 HPV types at various sequencing depths. The sensitivity progressively increased with higher sequence depth for both modes. At the depth of 2 reads/sample, the repeat-mask mode (76.8%) was much more sensitive than the homology-mask mode (29.4%) due to overlooking true HPV reads shared among different HPV types by the homology-mask mode (Fig. 3a). Sensitivity reached a plateau (>98.9%) at 50 reads for both modes. The specificity was ∼100% for both modes at 2-10 reads and was maintained for the homology-mask mode up to 1000 reads. However, the specificity progressively decreased at >50 reads and dropped to 89.8% at 1000 reads for the repeat-mask mode due to errors in genotyping of closely related HPV types (Fig. 3b). This is because that with increasing numbers of simulated HPV reads, additional reads are generated from homologous regions shared by different HPV types. When a read from homologous regions is mapped equally well to multiple locations, Bowtie2 randomly assigns one of the best mapped regions, so false positives of the tools are created when simulated reads from one HPV type (targeted type) are assigned to other HPV types (non-targeted types). Therefore, increasing sequencing depth will increase the chance of falsely detecting a non-targeted type in a sample or FP in the formula for calculating the specificity (see Materials and methods). In contrast, the currently available software HPVDetector (Chandrani et al., 2015) was less specific than both modes and less sensitive than the repeat-masked mode (Fig. 3).
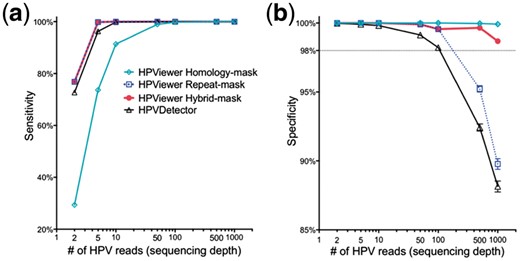
Evaluation of HPViewer using simulated HPV shotgun sequencing data. We compared the performance of three modes of HPViewer and HPVDetector using 100 100 simulated HPV samples. (a, b) Comparison sensitivity and specificity of HPViewer three modes and HPVDetector using simulated single HPV data with different sequencing depths: 2, 5, 10, 50, 100, 500, 1000 reads. Under each sequencing depth, we simulated each HPV type 100 samples. Considering HPVDetector only contained 143 types of HPV genomes, we only generated simulated reads from those HPV genomes
To surmount the low sensitivity of the homology-mask mode and the low specificity of the repeat-mask mode, we created a novel hybrid approach by combining the two modes using the pair-wise homology distance matrix. In this approach, the repeat-mask mode was used first to screen all HPV reads in a sample. If only a single type HPV was detected, the detected HPV was considered as true positive. If multiple HPV types were detected, their homology distance was determined using the pair-wise homology distance matrix. A HPV matching with no close relatives (homology distance > 0.35) was counted as true positive while closely related HPV types were examined with the homology-mask mode. Only HPV types re-detected using the homology-mask mode were considered as true positive (Fig. 4). The hybrid-mask mode had the same sensitivity of repeat-mask mode, 76.8%, at the depth of 2 reads and improved the specificity of the repeat-mask mode to 98.7% from 89.8% at 1000 reads (Fig. 3). These findings suggest that this hybrid-mask is optimal for detection of HPV in samples that contain either high or low number of HPV reads. This hybrid screening method was set as the default in the distributed version of HPViewer software.
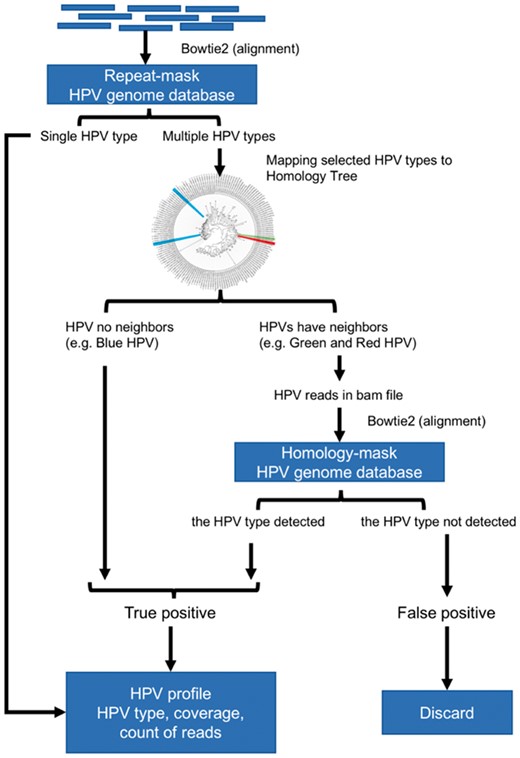
The workflow of hybrid-mode of HPViewer. The hybrid mode of HPViewer is a combination of repeat-mask database and homology-mask database through the homology distance matrix. The input is trimmed fastq file and the output is a table containing HPV types and abundance
3.5 Comparisons of the performance of HPViewer with HPVDetector, VirusTAP and Vipie using shotgun sequencing data of recurrent respiratory papillomatosis
We evaluated HPViewer with specimens infected by known HPV types. We performed shotgun sequencing on tumor tissues and matched oral washes from six patients with recurrent respiratory papillomatosis, known to be caused by HPV6 or 11. HPViewer detected HPV6 in 4 tumor tissues and 2 matched oral wash samples and HPV11 in 2 tumor tissues. In contrast, HPVDetector, a standalone program designed to directly genotype HPV reads from raw shotgun sequences, misidentified repeat reads from the human genome as false positive HPV19 (n = 4 samples), HPV71 (n = 9), or HPV82 (n = 7) (Table 2). HPVDetector also misidentified two reads as HPV 11 in sample 7 T which matched perfectly to HPV6. For these 12 samples, HPVDetector predicted an average of 1.9 wrong HPV types per sample (Fig. 5a). In the tumor tissues, HPVDetector consistently underestimated HPV read counts compared to HPViewer (P = 0.028, two-tailed paired t-test), for both HPV6 and HPV11 (Table 2).
Comparison of HPViewer and other programs on detection and genotyping of HPV in recurrent respiratory papillomatosis
| Sample ID | HPV type: number of reads detected | ||||
|---|---|---|---|---|---|
| Bowtie2 with complete | HPViewer | HPVDetector | VirusTAP | Vipie | |
| HPV genomes | |||||
| 3T |
| HPV6: 2721 | HPV6: 2150 | HPV6 | HPV6 |
| 7T |
| HPV6: 361 |
| No hits | HPV6 |
| 8T | HPV6: 1250 | HPV6: 1194 |
| HPV6 | HPV6 |
| 9T | HPV11: 4514 | HPV11: 4229 |
| HPV11 | HPV11 |
| 12T |
| HPV11: 228 |
| No hits | HPV11 |
| 15T | HPV6: 1285 | HPV6: 1223 |
| HPV6 | HPV6 |
| 3W |
|
|
| No hits | No hits |
| 7W |
| HPV6: 2 |
| No hits | No hits |
| 8W |
| No hits |
| No hits | No hits |
| 9W |
| No hits |
| No hits | No hits |
| 12W |
| No hits |
| No hits | No hits |
| 15W |
| No hits |
| No hits | No hits |
| Sample ID | HPV type: number of reads detected | ||||
|---|---|---|---|---|---|
| Bowtie2 with complete | HPViewer | HPVDetector | VirusTAP | Vipie | |
| HPV genomes | |||||
| 3T |
| HPV6: 2721 | HPV6: 2150 | HPV6 | HPV6 |
| 7T |
| HPV6: 361 |
| No hits | HPV6 |
| 8T | HPV6: 1250 | HPV6: 1194 |
| HPV6 | HPV6 |
| 9T | HPV11: 4514 | HPV11: 4229 |
| HPV11 | HPV11 |
| 12T |
| HPV11: 228 |
| No hits | HPV11 |
| 15T | HPV6: 1285 | HPV6: 1223 |
| HPV6 | HPV6 |
| 3W |
|
|
| No hits | No hits |
| 7W |
| HPV6: 2 |
| No hits | No hits |
| 8W |
| No hits |
| No hits | No hits |
| 9W |
| No hits |
| No hits | No hits |
| 12W |
| No hits |
| No hits | No hits |
| 15W |
| No hits |
| No hits | No hits |
Note: True positive and true negative results are in bold, and false positive results in italic and false negative results in underline.
Comparison of HPViewer and other programs on detection and genotyping of HPV in recurrent respiratory papillomatosis
| Sample ID | HPV type: number of reads detected | ||||
|---|---|---|---|---|---|
| Bowtie2 with complete | HPViewer | HPVDetector | VirusTAP | Vipie | |
| HPV genomes | |||||
| 3T |
| HPV6: 2721 | HPV6: 2150 | HPV6 | HPV6 |
| 7T |
| HPV6: 361 |
| No hits | HPV6 |
| 8T | HPV6: 1250 | HPV6: 1194 |
| HPV6 | HPV6 |
| 9T | HPV11: 4514 | HPV11: 4229 |
| HPV11 | HPV11 |
| 12T |
| HPV11: 228 |
| No hits | HPV11 |
| 15T | HPV6: 1285 | HPV6: 1223 |
| HPV6 | HPV6 |
| 3W |
|
|
| No hits | No hits |
| 7W |
| HPV6: 2 |
| No hits | No hits |
| 8W |
| No hits |
| No hits | No hits |
| 9W |
| No hits |
| No hits | No hits |
| 12W |
| No hits |
| No hits | No hits |
| 15W |
| No hits |
| No hits | No hits |
| Sample ID | HPV type: number of reads detected | ||||
|---|---|---|---|---|---|
| Bowtie2 with complete | HPViewer | HPVDetector | VirusTAP | Vipie | |
| HPV genomes | |||||
| 3T |
| HPV6: 2721 | HPV6: 2150 | HPV6 | HPV6 |
| 7T |
| HPV6: 361 |
| No hits | HPV6 |
| 8T | HPV6: 1250 | HPV6: 1194 |
| HPV6 | HPV6 |
| 9T | HPV11: 4514 | HPV11: 4229 |
| HPV11 | HPV11 |
| 12T |
| HPV11: 228 |
| No hits | HPV11 |
| 15T | HPV6: 1285 | HPV6: 1223 |
| HPV6 | HPV6 |
| 3W |
|
|
| No hits | No hits |
| 7W |
| HPV6: 2 |
| No hits | No hits |
| 8W |
| No hits |
| No hits | No hits |
| 9W |
| No hits |
| No hits | No hits |
| 12W |
| No hits |
| No hits | No hits |
| 15W |
| No hits |
| No hits | No hits |
Note: True positive and true negative results are in bold, and false positive results in italic and false negative results in underline.
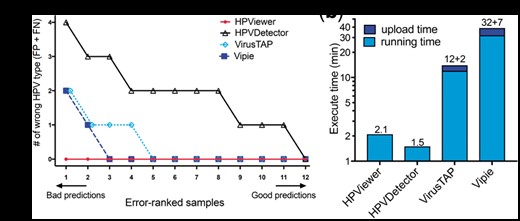
Comparison of different tools on recurrent respiratory papillomatosis shotgun sequencing samples. (a) Number of wrong predicted HPV types with respect to 12 shotgun sequencing samples. Wrong predicted HPV types consisted of false positive and false negative types. (b) Comparisons of execute time between HPViewer, HPVDetector, VirusTAP and Vipie for a fastq file containing 7.6M reads. VirusTAP and Vipie were web-based tools, so they also had upload time
VirusTAP is a web-based tool (Yamashita et al., 2016) that filters human and bacterial reads and utilizes de no assembly of filtered reads into contigs. It could only detect HPV from samples with very large number of HPV reads. For example, it was able to detect HPV6 in sample 15T in which HPViewer identified 1223 HPV reads but failed to detect HPV6 in samples 7T and 12T in which HPViewer identified 361 and 228 HPV reads. Vipie is another virus detection program (Lin et al., 2017) that utilizes de novo assembly of all reads into contigs. It was more sensitive than VirusTAP and had equivalent performance with HPViewer in tumor samples with >100 HPV reads. It successfully detected HPV6 in four tumor samples and HPV11 in two tumor samples. However, it failed to detect HPV in the oral wash samples that contained only a very small number of HPV reads. For example, the 6 HPV51 reads and 2 HPV6 reads in sample 3W and the 2 HPV6 reads in sample 7W were not observed by Vipie.
We compared the computing time of HPViewer with HPVDetector, VirusTAP and Vipie on analysis of a pair-end fastq file of sample 3T (fastq.gz file, 340 MB, 7.6M reads). The task took approximately two min for HPViewer and HPVDetector, 12 min for VirusTAP (plus 2 min uploading time) and 32 min for Vipie (plus 7 min uploading time) to complete (Fig. 5b). VirusTAP and Vipie cost longer time than HPViewer and HPVDetector to complete the same task because they needed extra time for the process of de novo assembly. VirusTAP pre-selects virus reads before the de novo assembly on a small number of selected sequences while Vipie performs de novo assembly on all reads before identifying HPV contigs. The longer time that Vipie needed than VirusTAP to analyze sample 3T reflects the fact that its scale of de novo assembly was much larger than that of VirusTAP.
3.6 Evaluation of HPViewer with shotgun sequencing data from healthy human subjects in the Human Microbiome Project
To evaluate the performance of HPViewer with datasets with unknown HPV status, we downloaded HMP Illumina metagenomic datasets that were originally generated from 1573 samples collected from 18 different body sites in healthy Americans. HPViewer detected 104 HPV types representing 4 HPV genera (Alpha, Beta, Gamma, Mu) (Eom et al., 2004) in 175 samples (Fig. 6 and Table 3) in 16 of the 18 body sites (overall prevalence: 11.10%). Of the 104 HPV types detected, 84 types of HPV (81.73%) should not be detectable by the widest spectrum cervical HPV detection kit, The Linear Array® (37 types of HPV) (Abreu et al., 2012). Among the 104 HPV types, the top four most commonly detected types were HPV51, 17, 18, 89 while 10 types of high-risk (66.67%, 10/15) and 7 types of low-risk (58.33%, 7/12) HPV were detected among these healthy samples. The body site with the highest prevalence of HPV was left retroauricular crease (65.22%), followed by right retroauricular crease (54.84%), vaginal inroitus (50.00%), anterior nares (37.96%) and posterior fornix (32.41%) while HPV was not detectable in samples from palatine tonsil and blood. According to their profiles of HPV prevalence, tongue dorsum, buccal mucosa, supragingical plaque, ileal pouch and stool were clustered as one group, and three vagina-related body sites—mid vagina, vaginal introitus, posterior fornix—were clustered together and three skin-related body sites—anterior nares, and left, right retroarticular crease—were clustered as another group (Fig. 6; Supplementary Table S3). It indicated that HPV prevalence was associated with its habitat environment and supported previous studies that suggested gut, mouth and skin have their own HPV diversity spectrums (Antonsson et al., 2000; Bottalico et al., 2011; Bzhalava et al., 2015b; Castro et al., 2012).
Summary of HPV-positive samples from the HMP
| Body site | Samples | HPV prevalence | HPV types |
|---|---|---|---|
| Total | 1573 | 11.13% | 104 |
| Anterior nares | 137 | 37.96% | 54 |
| Posterior fornix | 108 | 32.41% | 33 |
| Left retroauricular crease | 23 | 65.22% | 30 |
| Right retroauricular crease | 31 | 54.84% | 28 |
| Buccal mucosa | 184 | 6.52% | 11 |
| Tongue dorsum | 221 | 5.43% | 7 |
| Vaginal inroitus | 8 | 50.00% | 5 |
| Stool | 321 | 2.80% | 7 |
| Mid vagina | 20 | 25.00% | 5 |
| Ileal pouch | 34 | 8.82% | 1 |
| Nasopharynx | 162 | 0.62% | 2 |
| Keratinized gingiva | 14 | 7.14% | 1 |
| Palatine tonsil | 19 | 0.00 | 0 |
| Saliva | 7 | 14.29% | 3 |
| Subgingival plaque | 19 | 5.26% | 1 |
| Supragingival plaque | 210 | 2.86% | 6 |
| Throat | 13 | 7.69% | 1 |
| Blood | 42 | 0.00 | 0 |
| Body site | Samples | HPV prevalence | HPV types |
|---|---|---|---|
| Total | 1573 | 11.13% | 104 |
| Anterior nares | 137 | 37.96% | 54 |
| Posterior fornix | 108 | 32.41% | 33 |
| Left retroauricular crease | 23 | 65.22% | 30 |
| Right retroauricular crease | 31 | 54.84% | 28 |
| Buccal mucosa | 184 | 6.52% | 11 |
| Tongue dorsum | 221 | 5.43% | 7 |
| Vaginal inroitus | 8 | 50.00% | 5 |
| Stool | 321 | 2.80% | 7 |
| Mid vagina | 20 | 25.00% | 5 |
| Ileal pouch | 34 | 8.82% | 1 |
| Nasopharynx | 162 | 0.62% | 2 |
| Keratinized gingiva | 14 | 7.14% | 1 |
| Palatine tonsil | 19 | 0.00 | 0 |
| Saliva | 7 | 14.29% | 3 |
| Subgingival plaque | 19 | 5.26% | 1 |
| Supragingival plaque | 210 | 2.86% | 6 |
| Throat | 13 | 7.69% | 1 |
| Blood | 42 | 0.00 | 0 |
Summary of HPV-positive samples from the HMP
| Body site | Samples | HPV prevalence | HPV types |
|---|---|---|---|
| Total | 1573 | 11.13% | 104 |
| Anterior nares | 137 | 37.96% | 54 |
| Posterior fornix | 108 | 32.41% | 33 |
| Left retroauricular crease | 23 | 65.22% | 30 |
| Right retroauricular crease | 31 | 54.84% | 28 |
| Buccal mucosa | 184 | 6.52% | 11 |
| Tongue dorsum | 221 | 5.43% | 7 |
| Vaginal inroitus | 8 | 50.00% | 5 |
| Stool | 321 | 2.80% | 7 |
| Mid vagina | 20 | 25.00% | 5 |
| Ileal pouch | 34 | 8.82% | 1 |
| Nasopharynx | 162 | 0.62% | 2 |
| Keratinized gingiva | 14 | 7.14% | 1 |
| Palatine tonsil | 19 | 0.00 | 0 |
| Saliva | 7 | 14.29% | 3 |
| Subgingival plaque | 19 | 5.26% | 1 |
| Supragingival plaque | 210 | 2.86% | 6 |
| Throat | 13 | 7.69% | 1 |
| Blood | 42 | 0.00 | 0 |
| Body site | Samples | HPV prevalence | HPV types |
|---|---|---|---|
| Total | 1573 | 11.13% | 104 |
| Anterior nares | 137 | 37.96% | 54 |
| Posterior fornix | 108 | 32.41% | 33 |
| Left retroauricular crease | 23 | 65.22% | 30 |
| Right retroauricular crease | 31 | 54.84% | 28 |
| Buccal mucosa | 184 | 6.52% | 11 |
| Tongue dorsum | 221 | 5.43% | 7 |
| Vaginal inroitus | 8 | 50.00% | 5 |
| Stool | 321 | 2.80% | 7 |
| Mid vagina | 20 | 25.00% | 5 |
| Ileal pouch | 34 | 8.82% | 1 |
| Nasopharynx | 162 | 0.62% | 2 |
| Keratinized gingiva | 14 | 7.14% | 1 |
| Palatine tonsil | 19 | 0.00 | 0 |
| Saliva | 7 | 14.29% | 3 |
| Subgingival plaque | 19 | 5.26% | 1 |
| Supragingival plaque | 210 | 2.86% | 6 |
| Throat | 13 | 7.69% | 1 |
| Blood | 42 | 0.00 | 0 |

HPV prevalence summary of shotgun metagenomic data from HMP. 11 of 18 sites that were evaluated at least had two HPV positive samples. Sites are clustered vertically by their HPV prevalence pattern. The number in the parenthesis close to the body site label is the overall HPV positive sample/total samples and the number in the plot is the HPV prevalence for each HPV type in each body site
Co-occurrence of multiple HPV types in one sample were common with distinct patterns with respect to body sites (Fig. 7; Supplementary Table S4). In the co-occurrence network, there were 89 types of HPV co-occurring with others at least once. This network shared some similarity with previous study (Ma et al., 2014). Interestingly, HPV23-173 in skin, HPV54-89, and HPV39-51 in vagina were three most commonly observed co-occurrences (three times) and we did not find any co-occurrence relation shared between skin and vagina. These findings confirm that HPViewer is a broad range detection tool suitable for the evaluation of HPV presence beyond the female genital system.
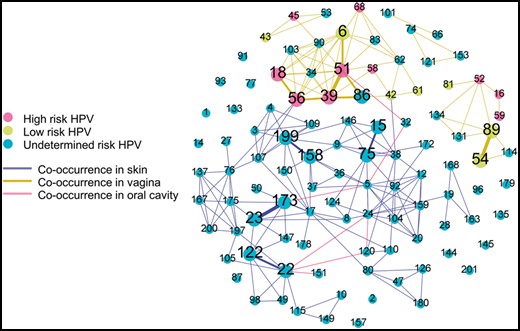
Co-occurrence graph of HPV in skin, vagina and oral cavity HMP samples. It consists of all 104 types of HPV. Each node represents one type of HPV and each edge represents the linked two nodes were found to co-existed. The thickness represents the frequency of co-occurrence in the range of 1–3. The nodes without any edges were not observed to have any co-occurrence. The skin includes anterior nares, left/right retroauricular crease; vagina includes mid vagina, posterior fornix, vaginal inroitus; oral cavity includes saliva, tongue dorsum, nasopharynx and buccal mucosa. Most co-occurrence (edges) happened in skin or vagina and there were only six co-occurrences in the oral cavity
4 Discussion
HPV is an important human pathogen not only because it is the main cause of cervical, oropharyngeal and anal cancers but also because of the increasing evidence to suggest non-cervical HPV types might play an etiological role for cancers of many other body sites. Given the inadequacy of cervical HPV detection kits to cover all 210 HPV types, metagenomic shotgun sequencing has emerged as one of the most promising strategies for the detection of HPV in human samples. Now, we show that HPV not only shares a substantial amount of homologous sequences among different HPV types but also shares extensive simple repeats with human and some prokaryotes. With HPVDetector, a previously published software program specially designed for detecting HPV in metagenomic data, we found that the intra-HPV homologous sequences cause errors in HPV genotyping and the shared repeats of human or prokaryotes origin can be mistaken as HPV DNA, indicating a need to design a program for more accurate detection and genotyping of HPV.
A HPV type is defined if its major capsid L1 gene sequence is less than 90% similar to that of any other types (Bzhalava et al., 2015a; Calleja-Macias et al., 2005; de Villiers, 2013). In the present study, we found it is common that regions of one HPV type share high similarity (>90%) with other types despite their L1 genes share less than 90% similarity. In 28 types of HPV, the shared portions accounted greater than 50% of their genomes, which may result from the conservation of early proteins (Bravo and Alonso, 2007). These variations in similarity among HPV genomes make it difficult to create an operational threshold for accurate genotyping among HPV types using short reads generated from shotgun sequencing. Yet it is clinically important to accurately determine the type of HPV in each sample, since HPV types differ in their pathogenic properties. We created a type-specific database by removal of all regions that shared >90% similarity among HPV types from the HPV reference genomes (homology-mask). We used the type-specific database in HPViewer and demonstrated that the homology-mask mode of HPViewer can reduce misclassification of reads to less than 0.3%.
An ideal software program for detection and genotyping HPV from shotgun sequences should be both specific and sensitive. Some HPV types share simple repeats with the human genome and prokaryotic genomes. In the papillomatosis samples, HPVDetector misclassified TG repeats of human origin as HPV71. VirusTAP takes two steps to ensure specificity. One is to filter out reads that are shared between HPV and non-HPV organisms and the second one which is also applied by Vipie, is to build up a large de novo assembled contigs to minimize the impact of local non-specific regions. This approach is demonstrated to be most specific among all programs evaluated. However, the high specificity is achieved at a cost of lower sensitivity due to the failure to assemble of contigs with sufficient length when a sample contains few HPV reads. In the papillomatosis study, VirusTAP failed to detect HPV6 in tumor samples despite each sample containing hundreds of HPV16 reads. Vipie is more sensitive than VirusTAP but unable to detect HPV in samples that contain less than 10 HPV reads. In contrast, HPVDetector is sensitive but less specific because of false positives from reads shared between HPV and human and prokaryotes or among HPV types.
HPViewer detects HPV by directly matching reads to HPV-specific reference genomes without de novo assembly. It achieved similar specificity to VirusTAP and Vipie with the threshold established by HPV type-specific PCR and higher sensitivity capable of detecting HPV in samples with as few as two HPV reads. The importance of detecting low HPV reads was exemplified in the study of recurrent respiratory papillomatosis. VirusTAP failed to recognize HPV infection in two (33%) of the six tumor samples despite several hundreds of HPV reads in the datasets, making it inadequate for diagnose of HPV infection in clinical samples. Vipie was unable to detect the presence of HPV6 in two oral samples in which there were two HPV reads in each sample. Because these four reads belonged to the same strains in the corresponding papilloma tumor samples, failure to detect them might underestimate the potential transmissibility of HPV from recurrent respiratory papillomatosis through the oral route.
In summary, HPViewer is a new tool designed for broad range detection and genotyping of HPV in shotgun sequencing data from human samples. It has high sensitivity by directly detecting HPV from raw sequence reads. It eliminates false positives by masking simple repeats in the reference HPV genomes shared by human and prokaryotes and reduces mistyping of HPV reads by masking homologous sequences shared among different HPV types. To optimize the trade-off between sensitivity and specificity, the hybrid mode of HPViewer integrates these two kinds of masked HPV genomes using the pair-wise homology distance matrix.
What is more, it uses the least space for data storage and provides faster time for analysis of HPV in a sample compared with other software programs available. HPViewer also has a built-in function to calculate HPV genome coverage. HPViewer is implemented with python and operates in the Linux environment so it can easily be used to process large numbers of samples. It produces a table containing HPV types detected, the number of matching reads and their depth of coverage on reference HPV genomes, and a bam file containing short reads aligned to HPV genomes, which can be visualized in the Integrative Genomics Viewer (Thorvaldsdóttir et al., 2013). With the rapidly decreasing cost of shotgun sequencing, metagenomics has emerged as one of the most effective strategies for the detection of HPV in clinical samples. HPViewer is a sensitive and specific tool for use in the analysis of HPV infection.
Acknowledgements
We thank the Applied Bioinformatics Laboratories at NYU School of Medicine for providing bioinformatics support and assisting with data analysis. This work utilized computing resources at the High Performance Computing Facility at NYU Langone Medical Center. We also thank the Genome Technology Center for expert sequencing. This shared resource is partially supported by a Cancer Center Support Grant, P30CA016087, at the Laura and Isaac Perlmutter Cancer Center. ZP is staff physician at the Department of Veterans Affairs New York Harbor Healthcare System. The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the U.S. Department of Veterans Affairs or the United States Government.
Funding
This work was supported in part by grants R21DE025352 from the National Institute of Dental and Craniofacial Research, R01AI110372 from the National Institute of Allergy and Infectious Diseases, and R01CA204113 and U01CA182370 from the National Cancer Institute of the National Institutes of Health, and the Dustin Micha Harper Recurrent Respiratory Papillomatosis Research Grant from the American Society of Pediatric Otolaryngology.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}