





Abstract
It is highly desirable to assemble genomes of high continuity and consistency at low cost. The current bottleneck of draft genome continuity using the second generation sequencing (SGS) reads is primarily caused by uncertainty among repetitive sequences. Even though the single-molecule real-time sequencing technology is very promising to overcome the uncertainty issue, its relatively high cost and error rate add burden on budget or computation. Many long-read assemblers take the overlap-layout-consensus (OLC) paradigm, which is less sensitive to sequencing errors, heterozygosity and variability of coverage. However, current assemblers of SGS data do not sufficiently take advantage of the OLC approach.
Aiming at minimizing uncertainty, the proposed method BAUM, breaks the whole genome into regions by adaptive unique mapping; then the local OLC is used to assemble each region in parallel. BAUM can (i) perform reference-assisted assembly based on the genome of a close species (ii) or improve the results of existing assemblies that are obtained based on short or long sequencing reads. The tests on two eukaryote genomes, a wild rice Oryza longistaminata and a parrot Melopsittacus undulatus, show that BAUM achieved substantial improvement on genome size and continuity. Besides, BAUM reconstructed a considerable amount of repetitive regions that failed to be assembled by existing short read assemblers. We also propose statistical approaches to control the uncertainty in different steps of BAUM.
http://www.zhanyuwang.xin/wordpress/index.php/2017/07/21/baum
Supplementary data are available at Bioinformatics online.
1 Introduction
Genome assembly refers to the reconstruction of the genomic sequence from a collection of sequencing reads. The methods of genome assembly have been developed along the evolution of sequencing technologies and can be categorized into two major frameworks: the overlap-layout-consensus (OLC) paradigm (Batzoglou et al., 2002; Myers, 1995; Myers et al., 2000) and the de Bruijn graph (DBG) representation of k-mers (Idury and Waterman, 1995; Pevzner et al., 2001). The OLC methods were more robust to sequencing errors, heterozygosity and coverage variations across the genome and played a key role before the coming of the second generation sequencing (SGS) technologies (Metzker, 2010). The DBG-based methods became a practical and popular choice in the era of SGS (Bankevich et al., 2012; Butler et al., 2008; Gnerre et al., 2011; Luo et al., 2012; Maccallum et al., 2009; Peng et al., 2010; Schatz et al., 2010; Simpson et al., 2009; Zerbino and Birney, 2008), since the k-mer decomposition of reads and tracking of Eulerian paths greatly reduce the computational complexity.
A complete genome assembly pipeline involves other important steps, like scaffolding and gap closing. SSAKE (Short Sequence Assembly by progressive K-mer search and 3’ read Extension) (Warren et al., 2007), SSPACE (SSAKE-based Scaffolding of Pre-Assembled Contigs after Extension) (Boetzer et al., 2011) and OPERA (Gao et al., 2011) are stand-alone tools to link contigs into scaffolds. GapCloser (Luo et al., 2012) and GapFiller (Boetzer and Pirovano, 2012) are two widely used tools to close gaps in the scaffolds. Iterative Mapping and Assembly for Gap Elimination (IMAGE) (Tsai et al., 2010) iteratively performs contig extending and merging to improve the assembly continuity. PAGIT (post-assembly genome-improvement) (Swain et al., 2012) is an integrative pipeline that consists of several open-source programs (Assefa et al., 2009; Otto et al., 2010; 2011; Tsai et al., 2010), and it is most suitable for bacterial or small eukaryote genomes.
An intrinsic challenge to genome assembly is the uncertainty caused by the widespread repetitive regions across a genome. The uncertainty is particularly high for the SGS reads since repetitive regions could produce many identical or near-identical reads. These short reads complicate the structure of DBG, and the sequences under the same pattern of repetitive region are likely to collapse together (Treangen and Salzberg, 2011). Therefore, the resulting assembly of highly repetitive genomes can be highly fragmented or severely shorter than the actual size. Some advances in solving repetitive genome assembly were reported recently. InGAP-sf (Shi et al., 2017) aimed to improve scaffolding. It addressed the uncertainty issue caused by repetitive sequences using a new strategy based on the combination of direct link and paired link graphs.
A possible solution to repetitive uncertainty is the single-molecule real-time sequencing technologies (SMRT; Eid et al., 2009; Roberts et al., 2013) represented by Pacific BioSciences (PacBio) and Oxford Nanopore Technologies. SMRT can sequence unprecedented long reads that span certain repetitive regions. The assembly methods of SMRT reads drew much attention in recent years (Berlin et al., 2015; Chin et al., 2013; Koren et al., 2017; Phillippy, 2017; Xiao et al., 2017). However, the high cost of SMRT sometimes makes it unaffordable to obtain enough coverage that ensures accuracy, completeness and continuity, especially for large genomes (Chakraborty et al., 2016). Moreover, the high error rate requires additional computations and can impact the assembly quality (Sovic et al., 2016). In comparison, Illumina sequencing reads are still of great value for genome assembly due to its high accuracy and low cost. The recent work (Wick et al., 2017) aimed at resolving the issue by integrating Illumina short reads and the third generation long sequencing reads.
In this article, we propose an integrative approach By Adaptive Unique Mapping (BAUM) and local OLC to improve genome assembly based on SGS paired-end/mate-pair libraries. BAUM has two modules: (i) construction of the genome unique regions that are taken as the initial contigs and (ii) iterative assembly, in which scaffolds are built, and contigs are extended and merged, aiming to reconstruct the repetitive regions along the iterations. In this scheme, the repetitive regions are separated by the unique regions. The reads from repetitive regions can possibly gain their locations with certainty through their mates mapped to the unique regions.
The unique regions, which are the basis of BAUM, are obtained from a given assistant genome through adaptive unique mapping and filtration under several rules of uniqueness. The assistant genome can be a genome from a close species, or an assembly by another de novo assembler. In the former case, BAUM is a reference-assisted assembler; while in the latter case, BAUM improves the result of other assemblers.
The iterative assembly module differs from IMAGE, which also takes an iterative scheme, in several crucial aspects. First, BAUM builds scaffolds in every iteration, since the extension of contigs increases the chance of more contigs’ being further linked together. Second, BAUM uses the time-proven OLC approach for contig extension because it is more robust to sequencing errors, heterozygosity and depth variations across the genome. Although the computational complexity of OLC is quadratic with respect to the read number, all contigs are extended independently and the number of reads used in extending one single contig is not large. Third, instead of pooling the reads that are used to extend the adjacent two contigs together, BAUM extends each contig by itself. We propose a robust statistical approach to guide the merging of adjacent contigs. The false positive merging can therefore be reduced. In addition, BAUM makes further modifications to improve efficiency, like incorporating libraries of diverse medium insert-sizes (300-2k) in contig extension, and performing the extension of all contigs in parallel to reduce the elapsed time.
Instead of using k-mers, BAUM extends contigs using original reads. Thus repeats whose lengths are longer than k but shorter than the read length could possibly be resolved by BAUM but not by the k-mer method. Along the iterative assembly, the extended parts on contigs could overlap with repetitive regions. If different copies of the repetitive regions would have certain divergence, we can still obtain UM reads under more stringent mapping criterion so that further extension can be gained.
BAUM was tested by simulation studies based on an Escherichia coli genome. We further tested BAUM on a plant genome and a vertebrate genome: a wild rice Oryza longistaminata (Zhang et al. 2015) and a parrot Melopsittacus undulatus (Bradnam et al., 2013), which was used in Assemblathon 2 (Bradnam et al., 2013). We found that BAUM substantially improved the assembly continuity and a considerable portion of repetitive regions was recovered. Being used to improve the Allpaths-LG’s (Gnerre et al., 2011) result for rice, BAUM resulted in a 42% increase of genome size and this was near to the estimated size. Besides, BAUM obtained a 21% increase of genome size by applying the iterative assembly module to the Newbler’s (454-Life-Sciences, 2012) assembly based on 454 long reads.
2 Materials and methods
2.1 Overview of BAUM
The workflow of BAUM is illustrated in Figure 1a. The input of BAUM contains the SGS reads and an assistant genome. The operations can be divided into two loops. The left loop generates the initial contigs, and the right loop carries out the iterative assembly.
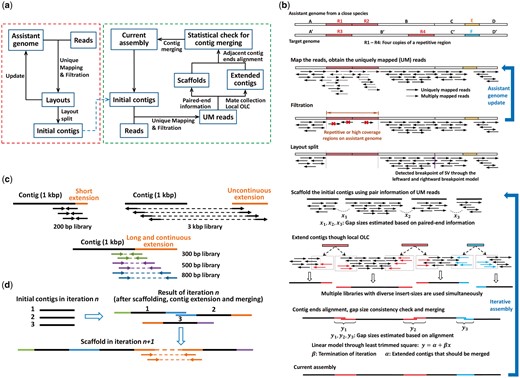
Illustration of BAUM. (a) Flowchart of operations. BAUM mainly consists of two loops shown, respectively, in the left and right rectangles. The left loop constructs the initial contigs by the UM reads with respect to an assistant genome. At the beginning of the loop, reads are mapped to the assistant genome and layouts of the UM reads are generated. A filtration is further performed based on two rules of uniqueness. The assistant genome is updated by the consensus of the layouts if its divergence to the target genome is fairly large, and reads are mapped again. This step can be performed for several iterations. After a step of layout split at the probable breakpoints of structural variations, the initial contigs are generated. In the right loop, scaffolds are built based on paired-end/mate-pair information, and those reads whose mates are UM to an end of a contig, are locally assembled using the OLC paradigm to extend the contig. The ends of adjacent contigs are aligned by Smith-Waterman algorithm to detect overlap, and a statistical approach is performed to decide which adjacent two extended contigs are merged. The resulting assembly in turn can serve as the basis for the next iteration. When BAUM is used to improve other assemblers’ results, the process can start from either the “Assistant genome” or “Current assembly”. In the latter case, only the right loop is performed. (b) An illustrative example explaining all steps in BAUM. (c) Multiple paired-end libraries with medium insert sizes can lead to longer and continuous contig extension. (d) In iterative assembly, contigs are extended in each iteration, and the extensions lead to more contigs’ being assembled into scaffolds in the next iteration (Color version of this figure is available at Bioinformatics online.)
At the beginning of the left loop (Fig. 1a), the reads are mapped to the assistant genome, and only the uniquely mapped (UM) reads are kept. The UM reads are further filtered by two uniqueness rules (Section 2.2). The resulted layouts are expected to correspond to the unique regions of the target genome, thus the uncertainty caused by repetitive regions is minimized at this stage. If the distance between the assistant and target genome is relatively large, the assistant genome is updated by the consensus of the layouts and the above procedures can be repeated for several rounds. Thereby the assistant genome gets closer to the target genome after this procedure. Due to the existence of structural difference between the two genomes, layouts are split at the probable breakpoints that are detected according to a statistical model we proposed, and the initial contigs are formed.
In the right loop (Fig. 1a), reads are mapped to the initial contigs, and scaffolds are built based on the paired-end/mate-pair information of the UM reads. Next, reads whose mates are UM to the ends of each contig are collected. Every contig, no matter whether it is linked with other contigs in the scaffold, is extended through local OLC approach. Libraries of multiple insert-sizes are pooled for contig extension.
After contig merging, we obtain the current assembly, and the resulting current assembly can be taken as a new assistant genome for the next iteration. After each iteration, the assembly is expected to cover more regions on the target genome and the contig/scaffold N50 is expected to grow (the contig/scaffold lengths are summed up successively from the longest to the shortest; when the sum reaches half of the total contig/scaffold length, the value of the corresponding contig/scaffold length is the contig/scaffold N50). When BAUM is used to improve other assembler’s result, the process can start from either the ‘assistant genome’ or ‘current assembly’ (Fig. 1a). In the latter case, only the right loop is performed.
We illustrate the BAUM method by an example in Figure 1b. The two blue arrows in Figure 1b correspond to the two loops in Figure 1a. The obtained initial contigs after the first loop include the four unique regions shared by the assistant and target genomes (A–D, A’–D’). The repetitive regions (R3 and R4) and the novel region (F) on the target genome are reconstructed through local OLC. We introduce the details of each step in Sections 2.2–2.7.
2.2 Adaptive unique mapping
UM reads are used in three different steps: (i) generate layouts of UM reads (Fig. 1a, left loop); (ii) construct scaffolds by UM mate-pairs (Fig. 1a, right loop); (iii) extend contigs by reads whose mates are UM (Fig. 1a, right loop). UM reads are defined by a mapping criterion and are obtained by a mapping algorithm. The mapping criterion needs to be selected adaptively in different steps. Any mapping tool designed for Second Generation Sequencing (SGS) reads (Langmead and Salzberg, 2012; Li and Durbin, 2009) can be used in this step. We adopt SEME (Sequential Exact seed-Match and Extend) (Chen et al., 2013) and define the mapping criterion by the maximal allowed mismatch number.
For the construction of layouts, if the inconsistency between the target and assistant genome is relatively large, particularly in the first iteration of the assistant genome update, we set a loose criterion to optimize the mapping rate and the UM rate. After updating (Fig. 1a, left loop), the assistant genome gets closer to the target, and we tighten the criterion accordingly in the following iterations. The mapping criterion in each iteration can be manually set by users or automatically set by BAUM. In the latter case, BAUM takes a small subset of reads and maps them under different parameter settings. The most stringent parameter setting that keeps the mapping rate above a certain threshold (default 80%) is selected as the mapping criterion. In each round of updating, we replace the assistant genome by the most frequent nucleotide in the layout at each position. For the sake of quality control, we only update the position if the coverage of the layout is higher than 6, and the maximal nucleotide frequency in the layout is higher than 60% (by default).
In scaffolding and contig extension, we do impose a relatively stringent criterion to minimize false positives. As contigs are extended, some may gradually overlap with the repetitive regions. Since different copies of the repetitive regions could have certain divergence, we can still obtain UM reads under more stringent mapping criterion so that further extension can be gained (Supplementary Note S1).
2.3 Filtration
After the layouts of UM reads are generated, the following kinds of UM reads are further filtered out: (i) UM reads that fall out of the unique regions on the assistant genome; (ii) UM reads that locate in the regions with higher than expected depth. The unique regions on the assistant genome are defined through ‘self-mapping’ (Supplementary Note S2). The filtration step aims to further filter out those reads that are generated from the repetitive regions on the target genome, thus reducing the uncertainty in assembly.
2.4 Layout split
It is necessary to detect the breakpoints of probable structural variations (SVs) between the assistant and target genome and split the layouts at the detected sites, thus the misassemblies at the structural level can be avoided. We simplify various kinds of SVs into two cases: leftward breakpoint and rightward breakpoint and establish a statistical model for breakpoint detection (Supplementary Note S3). Specifically, for each base on the assistant genome, we consider two null hypotheses, i.e. leftward breakpoint and rightward breakpoint. Then we carry out two statistical tests. The layout at the position is split if one of the tests fails to be rejected. We note that the primary control is the rate of false positives in which necessary splits are missed. Although the over-protection may lead to more false splits, they can be saved in the scaffolding and extension steps. The initial contigs are generated after layout split.
2.5 Iterative scaffolding and contig extension
Using the initial contigs as the basis, we iteratively carry out scaffolding and contig extension (Fig. 1a, right loop). Currently we use SSPACE (Boetzer et al., 2011) (version 3.0) and PHRAP (Green et al., 1994) (version 1.090518), respectively, for scaffolding and contig extension. The TAB file required by SSPACE is made according to the mapping information of UM reads, and the mapping step embedded in SSPACE is skipped.
BAUM applies OLC to only a relatively small subset of reads in each contig extension. As these reads’ mates are UM to the end of the contig, each subset is practically from a local region of the genome, and we term it as the local OLC. Since the extensions of all contigs are independent of each other, we can implement local OLC through parallel computation. The time complexity is linear with respect to the number of contigs and is quadratic only with respect to the sequencing coverage. The technical details are as follows.
Proposition: Denote the read length by , the number of contigs by and the sequencing coverage by . Then the average time complexity of local OLC for contig extension is .
A proof is given in Supplementary Note S5. It should be noticed that BAUM is able to use the read libraries of multiple insert sizes simultaneously in the contig extension. The libraries of medium insert sizes may lead to longer and continuous extensions (Fig. 1c). Moreover, contigs are extended in each iteration, and the extensions can lead to more contigs’ being assembled into scaffolds (Fig. 1d). Therefore, BAUM builds the scaffold in each iteration to improve continuity.
2.6 Statistical criterion for contig merging
We apply Smith–Waterman algorithm (Smith and Waterman, 1981) to align the adjacent extended contigs. The existence of high scoring segments in an alignment suggests a possible overlap of the two adjacent contigs. An estimated size of the gap between two adjacent contigs can therefore be calculated according to the position of the overlap. On the other hand, SSPACE also estimates the same gap distance using the insert-sizes of mate-pairs that are UM to the neighboring contigs. Statistically, we pool the two estimates for the cases of ‘no hanging end’ (Supplementary Fig. S1) and fit a linear regression line by the least trimmed squares (LTS) approach (Li, 2005). The adjacent extended contigs are merged for the cases whose residuals are within a certain range (Supplementary Note S4).
2.7 Stopping rule of assistant genome update and iterative assembly
In each round of assistant genome update, we map a small set of reads to the assistant genome under various criteria and select the most stringent parameter setting that keeps the mapping rate above a threshold (80% by default, Section 2.1). If the mismatch threshold in the selected parameter setting is no larger than 2 (default in BAUM), then we stop the assistant genome update; otherwise we continue the updating. The maximal number of assistant genome update is 10 by default.
For iterative assembly, at least two iterations are carried out. The iteration is terminated if at least one of the following conditions occurs: (i) the absolute deviation of the regression line slope from 1 is larger than a threshold (default value is 0.1) and (ii) the change of total contig length compared with the previous iteration is smaller than a given threshold (default value is 0.1%). The users can continue to run more iterations manually when necessary.
2.8 Illumina sequencing data
We test BAUM on two eukaryote genomes: O. longistaminata, a wild rice from Africa and M. undulatus. The data of O. longistaminata contain seven Illumina paired-end/mate-pair libraries of insert sizes—300, 400, 900, 2k, 5k, 10k and 20k (Zhang et al., 2015). The depth of the cleaned data is 230. The data of M. undulatus are from Assemblathon 2 (Bradnam et al., 2013), which contains 19 Illumina paired-end/mate pair libraries of insert sizes—220, 500, 800, 2k, 5k, 10k, 20k and 40k. The estimated depth is 289.
2.9 Validation by assembly from O. longistaminata 454 reads
An independent Roche 454 sequencing data (Zhang et al., 2015) of O. longistaminata is used to evaluate the accuracy of BAUM’s results. We assemble the 454 long reads using Newbler (version 2.9 20130529_1641). The statistics related to the assembly can be found in Table 2. Since long reads are likely to generate relatively more reliable assembly result, we take the assembled 454-contigs as a reference and evaluate the consistency between them and the assemblies based on Illumina short reads. The degree of consistency can be taken as a measure of BAUM’s accuracy. Specifically, we align each scaffold (or contig) in the Illumina short read assemblies to the 454-contigs using basic local alignment search tool (Camacho et al., 2009) and calculate the lengths of high scoring pairs (HSPs), indels and hanging ends (Supplementary Fig. S1). Shorter indels and less hanging ends indicate higher consistency.
Improvement of BAUM on other assemblers' results
| Species | Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rice | Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 271 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| After BAUM | 29.5 | 429.9 | 294 | 309 | 2676 | 94 h/14 | 13 | 1382 (96.0%) | 0 | 4 | 18.3 | |
| Rice | Newbler | 2.9 | 3.0 | 263 | 263 | 143 270 | 3 h | 18 | 942 (65.4%) | – | – | – |
| After BAUM | 25.0 | 511.4 | 319 | 332 | 5121 | 35 h/5 | 13 | 1391 (96.6%) | – | – | – | |
| Bird | Allpaths-LG | 58.6 | 10 935 | 1124 | 1183 | 5890 | 4.3 d | 380 | 4037 (82.1%) | 3 | 7 | 13.2 |
| After BAUM | 222.0 | 22 965 | 1148 | 1164 | 2703 | 4.6d/4 | 50 | 4592 (93.4%) | 0 | 0 | 17.3 | |
| BCM-HGSC | 159.3 | 13 049 | 1177 | 1330 | 56 982 | ∼25 d | ∼400 | 4497 (91.5%) | 0 | 2 | 14.0 |
| Species | Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rice | Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 271 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| After BAUM | 29.5 | 429.9 | 294 | 309 | 2676 | 94 h/14 | 13 | 1382 (96.0%) | 0 | 4 | 18.3 | |
| Rice | Newbler | 2.9 | 3.0 | 263 | 263 | 143 270 | 3 h | 18 | 942 (65.4%) | – | – | – |
| After BAUM | 25.0 | 511.4 | 319 | 332 | 5121 | 35 h/5 | 13 | 1391 (96.6%) | – | – | – | |
| Bird | Allpaths-LG | 58.6 | 10 935 | 1124 | 1183 | 5890 | 4.3 d | 380 | 4037 (82.1%) | 3 | 7 | 13.2 |
| After BAUM | 222.0 | 22 965 | 1148 | 1164 | 2703 | 4.6d/4 | 50 | 4592 (93.4%) | 0 | 0 | 17.3 | |
| BCM-HGSC | 159.3 | 13 049 | 1177 | 1330 | 56 982 | ∼25 d | ∼400 | 4497 (91.5%) | 0 | 2 | 14.0 |
Notes: (1) Ctg and Scaf are the abbreviations of contig and scaffold. (2) ‘Rice’ and ‘Bird’ represent O. longistaminata and M. undulatus. (3) Scaffolds shorter than 1 kbp were removed for comparisons. (4) BAUM's running time is from multiple iterations as indicated after the slash sign. (5) The NGA50 is calculated in the restricted scope of the 100 longest 454 contigs in the Rice case and in the restricted scope of the fosmid clones provided by Assemblathon 2 in the Bird case, respectively. (6) The running time and memory peak for BCM-HGSC were from Assemblathon 2 (Bradnam et al., 2013).
Improvement of BAUM on other assemblers' results
| Species | Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rice | Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 271 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| After BAUM | 29.5 | 429.9 | 294 | 309 | 2676 | 94 h/14 | 13 | 1382 (96.0%) | 0 | 4 | 18.3 | |
| Rice | Newbler | 2.9 | 3.0 | 263 | 263 | 143 270 | 3 h | 18 | 942 (65.4%) | – | – | – |
| After BAUM | 25.0 | 511.4 | 319 | 332 | 5121 | 35 h/5 | 13 | 1391 (96.6%) | – | – | – | |
| Bird | Allpaths-LG | 58.6 | 10 935 | 1124 | 1183 | 5890 | 4.3 d | 380 | 4037 (82.1%) | 3 | 7 | 13.2 |
| After BAUM | 222.0 | 22 965 | 1148 | 1164 | 2703 | 4.6d/4 | 50 | 4592 (93.4%) | 0 | 0 | 17.3 | |
| BCM-HGSC | 159.3 | 13 049 | 1177 | 1330 | 56 982 | ∼25 d | ∼400 | 4497 (91.5%) | 0 | 2 | 14.0 |
| Species | Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rice | Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 271 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| After BAUM | 29.5 | 429.9 | 294 | 309 | 2676 | 94 h/14 | 13 | 1382 (96.0%) | 0 | 4 | 18.3 | |
| Rice | Newbler | 2.9 | 3.0 | 263 | 263 | 143 270 | 3 h | 18 | 942 (65.4%) | – | – | – |
| After BAUM | 25.0 | 511.4 | 319 | 332 | 5121 | 35 h/5 | 13 | 1391 (96.6%) | – | – | – | |
| Bird | Allpaths-LG | 58.6 | 10 935 | 1124 | 1183 | 5890 | 4.3 d | 380 | 4037 (82.1%) | 3 | 7 | 13.2 |
| After BAUM | 222.0 | 22 965 | 1148 | 1164 | 2703 | 4.6d/4 | 50 | 4592 (93.4%) | 0 | 0 | 17.3 | |
| BCM-HGSC | 159.3 | 13 049 | 1177 | 1330 | 56 982 | ∼25 d | ∼400 | 4497 (91.5%) | 0 | 2 | 14.0 |
Notes: (1) Ctg and Scaf are the abbreviations of contig and scaffold. (2) ‘Rice’ and ‘Bird’ represent O. longistaminata and M. undulatus. (3) Scaffolds shorter than 1 kbp were removed for comparisons. (4) BAUM's running time is from multiple iterations as indicated after the slash sign. (5) The NGA50 is calculated in the restricted scope of the 100 longest 454 contigs in the Rice case and in the restricted scope of the fosmid clones provided by Assemblathon 2 in the Bird case, respectively. (6) The running time and memory peak for BCM-HGSC were from Assemblathon 2 (Bradnam et al., 2013).
2.10 Data accessibility
The raw sequencing data for O. longistaminata are available in SRA database of NCBI under the accession numbers SRX1156187 and SRX1156186. The Roche 454 long reads for O. longistaminata are available under SRX1156057. The sequencing data for M. undulates are accessible from ERR244154 to ERR244163. The fosmid sequences for M. undulates can be downloaded from http://gigadb.org/dataset/100062.
2.11 Computing equipments
We assembled the O. longistaminata genome using a tower server with 48 logic cores (Intel Xeon CPU E5-2697 v2 @ 2.70GHz) and 378 GB DDR3 memory. We assembled the M. undulates genome using a rack server with 48 logic cores (Intel Xeon CPU E5-2680 v3 @ 2.50GHz) and 504 GB DDR4 memory.
3 Results
3.1 Simulation tests on E. coli genome assembly
We first tested BAUM’s performance on simulated data. Specifically, we generated SVs (including insertion, deletion, duplication and reversion) using StructURal Variant majorIty VOte (SURVIVOR) (version 1.0.1) (Jeffares et al., 2017) on the E. coli reference genome (strain K-12 MG1655) with the default parameters. Then we obtained three different target genomes by mutating the structurally variated genomes at the rate of 5%, 10% and 25% (the variant sites were also generated by SURVIVOR). Hundred-bp paired-end/mate-pair reads were sampled from the three target genomes using ART (version 2.5.8) (Huang et al., 2012) (Supplementary Table S1). We took the E. coli reference genome (strain K-12 MG1655) as the initial assistant genome and ran BAUM for the three cases. After assistant genome updating and iterative assembly, BAUM resulted in a single contig that totally covered the target genome in each of the three target genomes (Case 1 in Supplementary Table S1). Moreover, we evaluated the results by quality assessment tool for genome assemblies (QUAST) (version 4.5) (Gurevich et al., 2013). If break-points occur when aligning assembled contigs to a reference genome, QUAST breaks the scaffolds into aligned blocks and calculates the N50 of these blocks, which is the so-called NGA50 of the assembly. QUAST showed no misassemblies in our results and the corresponding genome fractions were all higher than 99.99%. The NGA50 of the three assemblies were all higher than 4.68 Mbp (Case 1 in Supplementary Table S1).
Next, directly from the original E. coli genome (K-12 MG1655), we generated 100-bp paired-end/mate-pair sequencing reads of the same depth as above using ART. The original E. coli genome contains quite a fraction of long and short repeated sequences (Blattner et al., 1997). We applied BAUM to the simulated sequencing reads using different assistant genomes. As a test, we first obtained initial contigs from the E. coli genome itself by the left loop in Figure 1a. Then after eight rounds of iterative assembly, BAUM reached one single scaffold, in which no misassembly occurred and the NGA50 and contig N50 were, respectively, 4.63 Mbp and 2.5 Mbp. Second, we generated initial contigs using the three simulated genomes with nucleotide divergence rates of 5%, 10% and 25% from the original E. coli genome on top of SVs (last paragraph). Still, BAUM could reconstruct the genome with NGA50 4.63 Mbp, 4.63 Mbp and 2.69 Mbp, respectively, with contig N50 2.50 Mbp, 2.50 Mbp and 0.61 Mbp, respectively, and without any misassembly (Case 2 in Supplementary Table S1). We also assembled the simulated reads using SOAPdenovo2 (version 2.04) plus GapCloser (version 1.12) and compared the result with those of BAUM. BAUM outperformed by less local misassemblies and single-scaffold continuity except the situation with 25% divergence rate (Case 2 in Supplementary Table S1).
We further used BAUM to improve the results of de novo assemblers. When we took the results from Allpaths-LG (version 52488) as the assistant genome, after six iterations of iterative assembly, BAUM reached three scaffolds, in which the scaffold N50, NGA50 and contig N50 were, respectively, 3.06 Mbp, 3.04 Mbp and 469 kbp without any misassembly (Case 3 in Supplementary Table S1). In comparison, the GapCloser based on the Allpaths-LG’s results ended up with 4 scaffolds and 11 misassemblies. GapCloser worked much better with SOAPdenovo2, and reached 4 scaffolds and 1.22 Mbp of contig N50, while 21 local misassemblies also occurred. We applied one iteration of BAUM (right loop) to this result, and it reached one single scaffold with all gaps filled and only 20 local misassemblies (Case 4 in Supplementary Table S1).
These simulation results demonstrated that BAUM could extend and merge scaffolds/contigs with high fidelity. Its performance was robust with respect to the reference genome either from a relatively close species or from the results obtained by a fair de novo assembler. BAUM still worked even if the divergence between the target and assistant genomes was as high as 25%. Moreover, BAUM offered extra margin of scaffold/contig extension beyond existing methods.
3.2 Reference-assisted assembly of O. longistaminata
The genome of the cultivated rice, Oryza sativa japonica (IRGSP-1.0), was taken as the assistant genome. The estimated genome size of O. longistaminata based on the cytometry test was 329 Mbp (Zhang et al., 2015). The O. longistaminata and the O. japonica were estimated to diverge from Oryza glaberrima about 1.9 and 0.6 million years ago (Zhang et al., 2015). The divergence between O. japonica and O. longistaminata was fairly large, therefore, we ran four rounds of assistant genome updating before we carried out nine rounds of iterative assembly.
3.2.1 Convergence of assistant genome updating
We took the proportion of the reads that could be successfully mapped to the assistant genome as a measure of the similarity between the assistant and target genome. The proportion of successfully mapped reads remained at roughly the same level along the updating process, even as mapping criteria got more and more stringent, namely, the maximal allowed mismatch number decreased from 20 in the first round to 10 in the fourth round (all the sequencing reads were between 90 and 100 bp) (Supplementary Table S2). This indicates that the assistant genome converged to the target genome through updating.
3.2.2 Evaluation of initial contigs
The initial contigs were obtained after the steps of filtration and layout split. First, we evaluated the uniqueness of initial contigs. We observed that no more than 0.04% of the reads were mapped to multiple places on the initial contigs, while the multiple mapping rates were between 14.8% and 18.0% for the assistant genome from last round of update (Supplementary Fig. S2a). This implied that the uncertainty caused by the repetitive regions was largely reduced in initial contigs.
Second, we evaluated the results of layout split by aligning the initials contigs to the 454-contigs. The results showed that most of the initial contigs had no or very short hanging ends (Batzoglou et al., 2002), which demonstrated that no severe structural mistake existed in the initial contigs after layout split (Supplementary Fig. S3). We observed an example of a 22-nucleotide deletion in O. longistaminata. The layout split method detected it successfully (Supplementary Fig. S2b).
3.2.3 Iterative assembly
The libraries of insert-size 300, 400 and 900 were used in all iterations of iterative assembly, and we also added the 2 kbp library from iteration 5. The contig N50 increased steadily from 947 bp of the initial contigs to 27 887 bp, a 28-fold increase. The final scaffold N50 was 500 737 bp. Correspondingly, the number of closed gaps had a decreasing trend, down from 188 107 in the first iteration to 507 in the last iteration. It is noticed that the total size of initial contigs was only 211 Mbp, while the final scaffold size was 301 Mbp (Fig. 2a–d, Supplementary Table S3). This demonstrates that the iterative assembly recovered about one-third of the target genome.
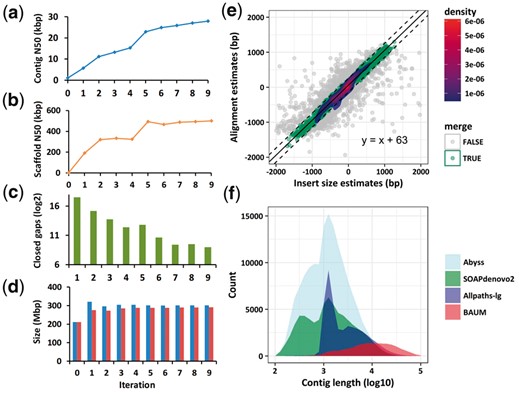
(a)–(d) Statistics of reference-assisted assembly of O. longistaminata. The x-axes represent the numbers of iteration, where ‘0’ represents the initial contigs. The statistics are: (a) contig N50, (b) scaffold N50, (c) number of closed gaps, (d) Total scaffold sizes (left bars) and total contig sizes (right bars). (e) Scatter plot of estimated gap sizes and the fit by LTS. It shows the case in the second round of iterative assembly. The x-axis is the estimate obtained through alignment of the two adjacent extended contigs. The y-axis is the estimate obtained using the insert-size of paired-end/mate-pair library in scaffolding step. A line of slope 1 is fit by LTS. The vertical distances between the dashed lines and the solid line are 200 bp and those dots falling inside the two dashed lines represent the adjacent extended contigs that are merged. (f) Comparison of contig length distribution between BAUM’s reference-assisted assembly result for O. longistaminata and three de novo assemblers (Color version of this figure is available at Bioinformatics online.)
3.2.4 Evaluation of iterative assembly
To evaluate the assembly accuracy, we checked the consistency by aligning the BAUM’s scaffolds to the 100 longest 454-contigs (Section 2.9). Among the 136 HSPs, only 1 had an obvious inconsistency (lengths of indel or hanging ends larger than 1 kbp) (Supplementary Table S4). Using these 454-contigs as the reference, QUAST found only five local misassemblies in the BAUM's result, the second best next to ABySS' (Table 1, the third and second column from the right).
Comparison of BAUM (reference-assisted version) with three de novo assemblers on O. longistaminata
| Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SOAPdenovo2 | 5.4 | 161.0 | 185 | 279 | 12 090 | 3 h | 67 | 1204 (83.6%) | 0 | 7 | 11.7 |
| ABySS | 3.3 | 14.8 | 299 | 320 | 48 617 | 16 h | 89 | 1283 (89.1%) | 0 | 3 | 8.9 |
| Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 010 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| Allpaths-LGa | 8.1 | 27.2 | 206 | 228 | 18 112 | 38 h | 138 | 1124 (78.1%) | 0 | 15 | 18.0 |
| BAUM | 27.9 | 500.7 | 291 | 301 | 14 947 | 142 h/9 | 13 | 1409 (97.8%) | 0 | 5 | 18.3 |
| Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SOAPdenovo2 | 5.4 | 161.0 | 185 | 279 | 12 090 | 3 h | 67 | 1204 (83.6%) | 0 | 7 | 11.7 |
| ABySS | 3.3 | 14.8 | 299 | 320 | 48 617 | 16 h | 89 | 1283 (89.1%) | 0 | 3 | 8.9 |
| Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 010 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| Allpaths-LGa | 8.1 | 27.2 | 206 | 228 | 18 112 | 38 h | 138 | 1124 (78.1%) | 0 | 15 | 18.0 |
| BAUM | 27.9 | 500.7 | 291 | 301 | 14 947 | 142 h/9 | 13 | 1409 (97.8%) | 0 | 5 | 18.3 |
Cheat mode of Allpaths-LG, in which the genome of O. sativa japonica was used to assist the assembly.
Notes: (1) Ctg and Scaf are the abbreviations of contig and scaffold, respectively. (2) Only the 2 kbp mate-pair library was used in Allpaths-LG’s scaffolding step because the program could not handle more than one mate-pair libraries in our server. (3) Since the Allpaths-LG did not output the scaffolds longer than 1 kbp, scaffolds shorter than 1kbp in all other assemblies were skipped for fair comparisons. (4) BAUM’s running time is from the last updated assistant genome to the final assembly result, with nine iterations of scaffolding, contig extension and merging. (5) The NGA50 is calculated in the restricted scope of the 100 longest 454 contigs.
Comparison of BAUM (reference-assisted version) with three de novo assemblers on O. longistaminata
| Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SOAPdenovo2 | 5.4 | 161.0 | 185 | 279 | 12 090 | 3 h | 67 | 1204 (83.6%) | 0 | 7 | 11.7 |
| ABySS | 3.3 | 14.8 | 299 | 320 | 48 617 | 16 h | 89 | 1283 (89.1%) | 0 | 3 | 8.9 |
| Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 010 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| Allpaths-LGa | 8.1 | 27.2 | 206 | 228 | 18 112 | 38 h | 138 | 1124 (78.1%) | 0 | 15 | 18.0 |
| BAUM | 27.9 | 500.7 | 291 | 301 | 14 947 | 142 h/9 | 13 | 1409 (97.8%) | 0 | 5 | 18.3 |
| Tool | Ctg N50 (kbp) | Scaf N50 (kbp) | Ctg size (Mbp) | Scaf size (Mbp) | Scaf number | Running time | Memory peak (Gb) | Complete BUSCOs | Mis- assemblies | Local misassemblies | NGA50 (kbp) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SOAPdenovo2 | 5.4 | 161.0 | 185 | 279 | 12 090 | 3 h | 67 | 1204 (83.6%) | 0 | 7 | 11.7 |
| ABySS | 3.3 | 14.8 | 299 | 320 | 48 617 | 16 h | 89 | 1283 (89.1%) | 0 | 3 | 8.9 |
| Allpaths-LG | 8.1 | 27.7 | 207 | 228 | 18 010 | 37 h | 138 | 1125 (78.1%) | 1 | 8 | 17.5 |
| Allpaths-LGa | 8.1 | 27.2 | 206 | 228 | 18 112 | 38 h | 138 | 1124 (78.1%) | 0 | 15 | 18.0 |
| BAUM | 27.9 | 500.7 | 291 | 301 | 14 947 | 142 h/9 | 13 | 1409 (97.8%) | 0 | 5 | 18.3 |
Cheat mode of Allpaths-LG, in which the genome of O. sativa japonica was used to assist the assembly.
Notes: (1) Ctg and Scaf are the abbreviations of contig and scaffold, respectively. (2) Only the 2 kbp mate-pair library was used in Allpaths-LG’s scaffolding step because the program could not handle more than one mate-pair libraries in our server. (3) Since the Allpaths-LG did not output the scaffolds longer than 1 kbp, scaffolds shorter than 1kbp in all other assemblies were skipped for fair comparisons. (4) BAUM’s running time is from the last updated assistant genome to the final assembly result, with nine iterations of scaffolding, contig extension and merging. (5) The NGA50 is calculated in the restricted scope of the 100 longest 454 contigs.
The reliability status of the iterations can partially be monitored by the scatter plot of the two estimates of gap sizes between adjacent contigs, one by alignment and one by insert sizes of mate pairs (Supplementary Fig. S4). Figure 2e shows this scatter plot in the second iteration. We fit a simple linear regression by LTS. The dots falling inside the band correspond to the cases of good consistency, while those falling out of the linear band correspond to the somewhat inconsistent cases. The inconsistency may be caused by misalignment in the presence of interspersed repeats or size errors in the libraries. The adjacent extended contigs were not merged for these occasions. When the slope estimate of LTS substantially deviates from 1 or the portion of good cases gets small, it suggests that iteration should be stopped.
3.2.5 Comparison with de novo assemblers
We compared the above reference-assisted assembly result with those generated by three widely used SGS assemblers: SOAPdenovo2 (Luo et al., 2012) (version 2.04), ABySS (Simpson et al., 2009) (version 1.9.0) and Allpaths-LG (Gnerre et al., 2011) (version 52488). It can be seen from Table 1 that the contig N50 of BAUM was 3-fold, 5-fold and 8-fold of that of Allpaths-LG, ABySS and SOAPdenovo2, respectively. The large proportion of repetitive regions on the rice genome accounts for the smaller N50 of the three de novo assemblers; whereas the design of BAUM resolves this problem to a great extent. It can be seen that the contig number of BAUM was significantly smaller than the others, and the length distribution of BAUM contigs had an obvious shift to the right compared with the others (Fig. 2f). All these indices demonstrate that BAUM achieved the best continuity. The genome size of the BAUM assembly was close to the estimated value given by the cytometry test (329 Mbp), while those corresponding to SOAPdenovo2 and Allpaths-LG were much smaller. We assessed the completeness of genome assembly using BUSCO (Benchmarking Universal Single-Copy Orthologs) (version v3) (Simão et al., 2015), which was a software that assessed the genome assembly and annotation completeness based on evolutionarily informed expectations of gene content). Table 1 shows the number of complete BUSCOs (predicted gene content) in these assemblies. It can be seen that BAUM achieved the best results, 1409 (97.8%). In other words, BAUM’s result was most complete from the perspective of single-copy orthologs.
3.3 Improvements of other assemblers’ results
To examine BAUM’s ability to improve other assemblers’ results, we ran BAUM on the Allpaths-LG’s assembly for O. longistaminata. We carried out 14 rounds of iterative assembly (Supplementary Table S5, Supplementary Fig. S5). The contig N50 grew from 8.1 kbp to 29.5 kbp, and the final contig size (non-N base number) increased by 42% (Table 2), indicating that BAUM recovered a considerable amount of unassembled regions. We further checked the consistency between the assembly and the top 100 longest 454-contigs as described above. Among the 126 HSPs (Supplementary Table S6), only three serious inconsistencies (lengths of indel or hanging ends larger than 1 kbp) were found.
As another example, we applied BAUM to Newbler’s assembly of 454 long reads, which we used above to evaluate the assembly results. The iterative assembly was carried out for five iterations (Supplementary Table S7, Supplementary Fig. S6). The genome size grew from 263 Mbp to 332 Mbp, and the contig N50 grew from 2.9 kbp to 25.0 kbp (Table 2). Moreover, the contig and scaffold numbers decreased by, respectively, 1 and 2 orders of magnitude. This indicates that BAUM can improve the assembly obtained from long reads too.
We also ran BAUM on the Allpaths-LG’s result for M. undulatus, a parrot from Australia. This genome was taken as a gage in Assemblathon 2 (Bradnam et al., 2013) to evaluate the performance of different assemblers. It was mentioned in Assemblathon 2 that the estimated genome size was 1.2 Gbp. We used the same parameters as in Assemblathon 2 to generate an assembly using Allpaths-LG, and then carried out four rounds of BAUM iterative assembly (Supplementary Table S8, Supplementary Fig. S7). Although the assembly result for Allpaths-LG was already satisfactory, BAUM made further improvement on continuity (Table 2). The contig N50 grew from 58 555 bp to 222 017 bp, a 3-fold increase, while the scaffold N50 grew from 17.9 Mbp to 23.0 Mbp.
To evaluate the accuracy of the result, we aligned the scaffolds before and after running BAUM to the 46 fosmid sequences provided by Assemblathon 2. The improvements made by BAUM were all validated by the fosmid sequences despite four small indels (Supplementary Table S9). Using these fosmid sequences as the reference, QUAST found no misassembly in BAUM's result (Table 2, the third and second column from the right).
Apart from contig N50 and genome size, the complete BUSCOs also increased by a great deal in these three cases. The number of complete BUSCOs for M. undulatus increased from 4037 (82.1%) to 4592 (93.4%), even better than that of BCM-HGSC, one of the best assemblies of the same genome in Assemblathon 2 which was obtained by integrating 454, Illumina and PacBio sequencing data (Table 2). Thus, on top of the state-of-art results, BAUM can make extra room for biology research.
3.4 Comparison of BAUM’s iterative assembly with GapCloser and IMAGE
When BAUM was used to improve Allpaths-LG’s assembly for O. longistaminata, we replaced the steps of contig extending and merging with GapCloser (Luo et al., 2012) in the beginning four iterations, and compared the results. The total size of contigs (the number of non-N bases) of BAUM increased steadily to 271 Mbp in the first four iterations, while that of GapCloser stopped around 246 Mbp (Supplementary Table S10). It is noted that BAUM extended each contig on both sides, even if it was located at the end of a scaffold. Larger contig sizes provided a higher chance of more contigs’ being linked in the next iteration. The memory usage for BAUM was much less than GapCloser, since the extension for all contigs were separate and the memory usage of extending one contig with local OLC approach was not that large (Supplementary Table 11).
We also compared the performance of BAUM with IMAGE (Tsai et al., 2010) (version 2.4.1) in the first iteration of the same case. The total size of contigs (the number of non-N bases) only increased from 207 Mbp to 210 Mbp for IMAGE, whereas it reached 238 Mbp for BAUM. The contig N50 and number of merged contigs for BAUM (11 224 bp and 11 520) were also greater than that of IMAGE (8340 bp and 1236), respectively. In terms of the time usage of contig extending and merging, the elapsed time for BAUM was 2h 22min, which was much less than that of IMAGE, 55h. It is noted that this version of IMAGE was not able to extend all contigs in parallel.
3.5 Reconstruction of repetitive regions
We took RepBase (Bao et al., 2015) to evaluate the repetitive region reconstruction. Specifically, the RepBase (version 22.11) library of Oryza (oryrep.ref) and ‘other vertebrates’ (vrtrep.ref) were used to evaluate the rice and parrot assemblies, respectively. We aligned the assemblies to the RepBase and labeled a region as a repetitive region if its similarity to the reference was higher than 70%. Figure 3a shows the results of the reference-assisted assembly of O. longistaminata. The proportion of repetitive regions in the initial contigs was only 5.2%, while it increased to 18.2% in the final assembly. Among the regions that were reconstructed through the iterative assembly, more than half (52.7%) were repetitive regions. These demonstrate that after unique mapping and filtration, BAUM reduced the uncertainty of repetitive regions in the initial contigs, and it recovered a considerable amount of repetitive regions in the following iterative assembly. Figure 3b shows the case of Allpaths-LG + BAUM on O. longistaminata. The genome size increased greatly after BAUM was applied, and about one-third (35.2%) of the extended regions were repetitive regions. In the case of M. undulatus (Fig. 3c), the proportion of repetitive regions in the extended sequences was 34.4%, which was significantly larger than that in the whole assembly (3.1%). All these results demonstrate that BAUM has a unique ability in reconstructing repetitive regions.
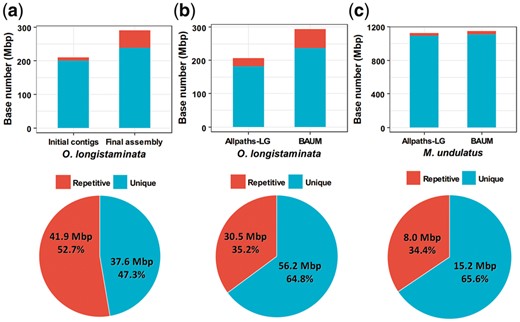
Reconstruction of repetitive regions in three applications of BAUM. The initial contigs (or the result of Allpaths-LG) and final assemblies are aligned to RepBase and a region is identified as a repetitive region if it can be aligned to the RepBase with identity higher than 70%. The bar charts show the lengths of repetitive and unique regions. The pie charts show the proportions of repetitive and unique regions in the extended parts. The RepBase library of Oryza is used to evaluate the two assemblies of O. longistaminata, while the RepBase library of ‘other vertebrates’ (vrtrep.ref) is used to evaluate the assembly of M. undulatus. (a) Reference-assisted assembly of O. longistaminata. (b) Improvement of Allpaths-LG’s assembly for O. longistaminata. (c) Improvement of Allpaths-LG’s assembly for M. undulatus (Color version of this figure is available at Bioinformatics online.)
This edge of BAUM over other assemblers is illustrated by three repetitive regions, namely the Gypsy-47_OS-LTR (Supplementary Fig. S8a), OLO24 (Supplementary Fig. S8b) and Helitron-N107B_OS (Supplementary Fig. S8c). These three cases were taken from the reference-assisted assembly of O. longistaminata. We can see that all the three assemblers, SOAPdenovo2, Allpaths-LG and Abyss, had problems in these regions, while BAUM successfully reconstructed the three repetitive regions together with their neighborhoods.
4 Discussion
In this article, we propose an approach BAUM that can perform reference-assisted assembly or improve the results by other assemblers. As shown by the cases of O. longistaminata and M. undulatus, BAUM greatly improved the continuity and genome size, even if the original assembly was highly fragmented and far from complete. Moreover, a considerable amount of repetitive regions, which failed to be assembled by the mentioned existing methods, were successfully reconstructed by BAUM. The performance of BAUM demonstrates that for genome assembly, the SGS data still have great value to be exploited.
The datasets in our experiments have a common feature: the paired-end libraries with medium insert-sizes (300-2k) have high coverage. Since BAUM pools paired-end libraries of diverse insert-sizes in contig extension, a longer continuous extension can be obtained within each iteration. This is crucial for improving the highly fragmented assembly. The above results indicate that generating multiple medium insert-size libraries with higher coverage could lead to much better assembly results.
Unique regions are the key to the control of uncertainty in all steps of BAUM. The notion of unique regions has been used in the DNA assembly since the era of Sanger sequencing (see Celera Assembler (Myers et al., 2000) and ARACHNE (Batzoglou et al., 2002)). In the context of SGS, the read lengths are short and the balance between uncertainty and uniqueness is different. BAUM uses UM reads to deal with uncertainty, and the specific mapping criterion decides whether a read is UM. We provide a theoretical foundation for uniqueness in this work (Supplementary Note S1), which can generally be used in computational genome research.
Computationally, BAUM adopts local OLC to extend the contigs. Without decomposing the reads into k-mers, OLC can achieve more robust contig extension. The computational complexity of pair-wise alignment in OLC is quadratic with respect to read number. By distributing the reads according to their UM mates, the OLC is carried out locally around the ends of each contig. The number of reads involved in the extension of each contig is not large, and consequently the computation complexity is acceptable. Since the extensions for all contig are independent, we implement them in parallel.
The challenging computational requirements imposed by current assembly projects of large genomes using high coverage of SGS reads include not only the CPU number but also memory. For example, it is written in the manual of Allpaths-LG that the estimated memory usage is generally 1.7 times of the size of the dataset (Computational Research and Development Group, 2013). Since BAUM performs iterative assembly, we can take only a fraction of reads in de novo assembly to ease the memory burden. Although the assembled contigs are less complete to cover the whole genome, the missed part can be recovered through the iterative assembly, as shown in the case of O. longistaminata. Along the iterations, reads that are mapped to the inner parts of contigs can be discarded, so that the mapping time in the following iterations becomes shorter and shorter.
In the current version of BAUM, the initial contigs are generated either from the genome of a close species or from a de novo assembler. Different initial contigs may lead to different results. This can be seen in Table 2, in which initial contigs were generated by Allpaths-LG using Illumina reads and by Newbler using 454 reads. The former has a longer contig N50 value while the latter has a longer genome size. How to integrate these results is an interesting topic in the future investigation. The method proposed in (Zhao et al., 2008) is a relevant technique solving such a problem.
As SMRT long reads become more and more widely used in genome assembly, BAUM can potentially be incorporated into hybrid assembly (Zimin et al., 2017). The improvement on continuity and the increase on genome size by BAUM can reduce the number of long reads that need to be de novo assembled, and thereby reduce the cost.
Acknowledgements
We thank Dr. Wen Wang and Dr. Yesheng Zhang at Kunming Institute of Zoology, Chinese Academy of Sciences (CAS) for providing us with the Illumina HiSeq and Roche 454 sequencing data of O. longistaminata. We thank Liang Li from Chinese Academy of Sciences for her help with the software development. We thank Dr. Meng Zou at the University of Iowa for his critical suggestions on the organization and writing of the manuscript.
Funding
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB13040600), the National Natural Science Foundation of China (grant no. 91530105, 91130008), the National Center for Mathematics and Interdisciplinary Sciences of the CAS and the Key Laboratory of Systems and Control of the CAS.
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Anqi Wang and Zhanyu Wang authors should be regarded as Joint First Authors.


{kind=link}
{kind=link}
{kind=link}