





Abstract
OmicsPrint is a versatile method for the detection of data linkage errors in multiple omics studies encompassing genetic, transcriptome and/or methylome data. OmicsPrint evaluates data linkage within and between omics data types using genotype calls from SNP arrays, DNA- or RNA-sequencing data and includes an algorithm to infer genotypes from Illumina DNA methylation array data. The method uses classification to verify assumed relationships and detect any data linkage errors, e.g. arising from sample mix-ups and mislabeling. Graphical and text output is provided to inspect and resolve putative data linkage errors. If sufficient genotype calls are available, first degree family relations also are revealed which can be used to check parent–offspring relations or zygosity in twin studies.
omicsPrint is available from BioConductor; http://bioconductor.org/packages/omicsPrint.
Supplementary data are available at Bioinformatics online.
1 Introduction
Increasingly, human studies involve the generation and analysis of multiple omics data for large groups of individuals (Baranzini et al., 2010; Bonder et al., 2017). These efforts require careful data management and quality control as in each step of laboratory protocols and sample-logistics there is the risk of introducing sample mix-ups. The resulting errors in data linkage reduce the power to detect biologically meaningful results (Buyske et al., 2009). The importance of this issue is widely recognized, particularly in the field of genetics (Abecasis et al., 2001; Pedersen and Quinlan, 2017). However, for the linkage of genetic data with other omics data types, fewer tools are available. Moreover, they rely on indirect measures of genotypes (e.g. the effect of quantitative trait loci) resulting in ambiguous assignments (Westra et al., 2011). Also, current tools do not provide functionality to perform analyses within an omics data type other than genetics to detect errors or family relations. Here, we present omicsPrint, a versatile method for the detection of data linkage errors and family relations in large-scale multiple omics studies. The method uses a classification approach on the basis of genotype calls derived from the different data types resulting in a clear distinction between verified relationships and errors due to sample mix-ups, mislabelings and, if sufficient genotype calls are available, first-degree family relations.
2 Implementation
Identity by state (IBS) is a genetic similarity measure that compares at a single locus the genotypes between two individuals and counts the number of alleles shared (0, 1 or 2). The IBS mean and variance, calculated for a set of genetic variants, can be used to identify relatedness between individuals (Abecasis et al., 2001). OmicsPrint applies linear discriminant analysis on the IBS means and variances for all individuals in a study to determine sample mix-ups and classify first degree family relations automatically obviating the need for arbitrary thresholds. Key to the comparisons across multiple types of omics data is the availability of a set of overlapping genetic variants. Specifically, for DNA methylation data obtained using the Illumina Human Methylation arrays (450 k/EPIC) an unsupervised clustering approach using the K-means algorithm was implemented to call genotypes for CpG-probes that are known to be affected by bi-allelic SNPs in probe-sequences (Zhou et al., 2017). Additionally, omicsPrint provides a subset of the annotation data generated by Zhou et al., (2017) to ease the extraction of CpG-probes affected by bi-allelic SNPs specificly for different populations. For RNA-sequencing data, several methods exist for the extraction of genotype calls (Piskol et al., 2013).
3 Example
To illustrate the use of our method, we performed sample relation verification within and across omics data types using multiple publicly available data sets. First, we used DNA sequencing from the 1000 genomes project (Birney and Soranzo, 2015) and Illumina 450k array data (GSE39672) (Moen, 2013) for 134 HapMap individuals. IBS mean-variance plots generated by omicsPrint revealed the expected clusters of unrelated samples (comparing a sample with all other samples) and related samples (sample with itself) for both genotypes measured using DNA sequencing and genotypes inferred from Illumina methylation array data using omicsPrint (Fig. 1A and B). Moreover, the IBS mean-variance of DNA-sequencing versus DNA methylation array data using 437 overlapping genotypes verified correct data linkage for all but 2 individuals (Fig. 1C). When artificially introduced, linkage errors can clearly be detected (Fig. 1D). A simulation indicated that ∼250 genotypes are sufficient to detect data linkage errors (Supplementary Material S1).
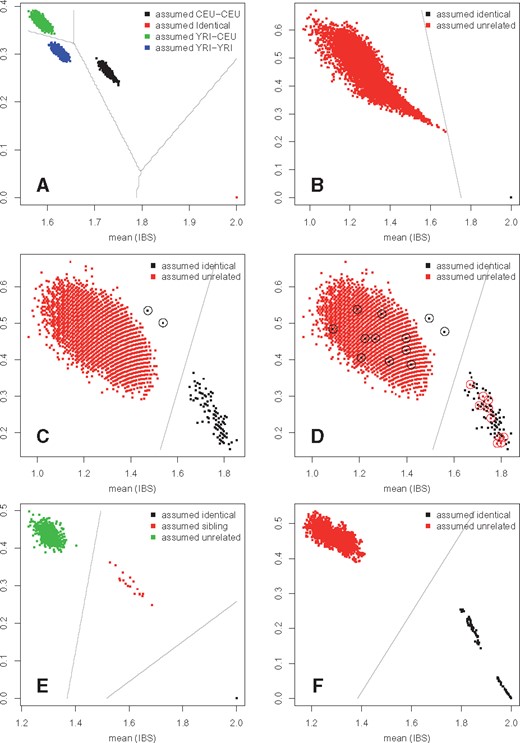
Graphical output of omicsPrint: IBS mean-variance plots of (A) DNA-seq/DNA-seq comparison for 104 HapMap individuals using 7107 genotypes extracted from 1000 Genomes data, (B) DNAm-array/DNAm-array comparison for 134 HapMap individuals using 8977 genotypes inferred from DNA methylation array data with omicsPrint, (C) DNA-seq/DNAm-array cross-omics comparison for 133 HapMap individuals using 437 genotypes overlapping between datasets, (D) same as (C) but now after introducing 10 artifical sample mix-ups detectable as differently colored dots in a cluster (two relations need further inspection as these appear in an unexpected region; black dots with circles). (E) DNAm-array/DNAm-array comparison for 18 sibling pairs using 1002 inferred genotypes and (F) DNAm-array/DNAm-array comparison for 2 tissue types sampled from 30 individuals using 895 inferred genotypes (Color version of this figure is available at Bioinformatics online.)
Second, we inferred genotypes from DNA methylation array data on 18 sibling pairs (GSE102177) (Kim et al., 2017) to show that our approach can reliably detect first degree family relationships (1002 genotypes; Fig. 1E). Likewise, these inferred genotypes can be used to determine the zygosity of twin pairs (Supplementary Material S2). Finally, genotypes inferred from DNA methylation data obtained on two tissues (dermis and epidermis) from 30 individuals (Vandiver et al., 2015) (GSE52980) to illustrate the utility of omicsPrint to match multiple samples from a single individual (895 genotypes; Fig. 1F).
4 Conclusion
We describe omicsPrint, a new software method for the reliable and fast detection of data linkage errors in large-scale multiple omics studies. The method uses genotype calls that are either measured or inferred from RNA-seq or DNA methylation array data. Automatic classification of genetic similarity based on IBS and supporting graphical and text output allows users to quickly review and resolve data linkage errors.
Funding
This work was supported by BBMRI-NL, a research infrastructure financed by the Dutch government [NWO 184.021.007 and NWO 184.033.111].
Conflict of Interest: none declared.
References


{kind=link}