





Abstract
The fundamental challenge of modern genetic analysis is to establish gene-phenotype correlations that are often found in the large-scale publications. Because lexical features of gene are relatively regular in text, the main challenge of these relation extraction is phenotype recognition. Due to phenotypic descriptions are often study- or author-specific, few lexicon can be used to effectively identify the entire phenotypic expressions in text, especially for plants.
We have proposed a pipeline for extracting phenotype, gene and their relations from biomedical literature. Combined with abbreviation revision and sentence template extraction, we improved the unsupervised word-embedding-to-sentence-embedding cascaded approach as representation learning to recognize the various broad phenotypic information in literature. In addition, the dictionary- and rule-based method was applied for gene recognition. Finally, we integrated one of famous information extraction system OLLIE to identify gene-phenotype relations. To demonstrate the applicability of the pipeline, we established two types of comparison experiment using model organism Arabidopsis thaliana. In the comparison of state-of-the-art baselines, our approach obtained the best performance (F1-Measure of 66.83%). We also applied the pipeline to 481 full-articles from TAIR gene-phenotype manual relationship dataset to prove the validity. The results showed that our proposed pipeline can cover 70.94% of the original dataset and add 373 new relations to expand it.
The source code is available at http://www.wutbiolab.cn: 82/Gene-Phenotype-Relation-Extraction-Pipeline.zip.
Supplementary data are available at Bioinformatics online.
1 Introduction
The biomedical literature is vast (Cohen and Hersh, 2005), and there is an urgent need to process publications automatically and mine embedded knowledge in the literature to create research hypotheses. Recently, biomedical relationship extraction has gained attention for many downstream text-mining applications, such as event extraction, database creation, knowledge discovery, question answering and decision-making. Natural language processing (NLP) systems have been used for mining special relationships from texts as protein–protein interactions (Papanikolaou et al., 2015; Yang et al., 2011; Zhu et al., 2015), genes and diseases (Coulet et al., 2010; Kim et al., 2017), drug–drug interactions (Segura Bedmar et al., 2011, 2013), as well as among genes, drugs and mutations (Cheng et al., 2008; Rindflesch et al., 1999). Such relationship extraction contributes to the development of pharmacogenomics, clinical trial screening and adverse drug reaction identification (Luo et al., 2017).
The central challenge of modern genetic analysis is to establish genotype–phenotype correlations (Cobb et al., 2013; Fu et al., 2014), which are often found in the biomedical literature, but the volume warrants an automatic and reliable system to extract these information from the text.
Although relationships have been identified among numerous biological entities, the system for extracting gene–phenotype relationships from the literature is very limited. Regarding species types, the current research focuses more on the relationships between human genes and phenotypes (Collier et al., 2015; Yang et al., 2015). To our knowledge, there is few such studies for plants. Regarding entity types, research on identifying specific phenotypes such as diseases and gene relationships has received great attention (Kim et al., 2017; Özgür et al., 2008; Singhal et al., 2016). However, text-mining systems that can recognize various phenotype and gene relationships are more difficult and are less robust. The system generally involves annotating raw text with named entities and extracting relationships between these entities. (Luo et al., 2017) Named entity recognition (NER) is the foundation of relationship extraction and the effect of entity recognition greatly affects relationship extraction results. (Chun et al., 2006) With gene–phenotype relationship extraction, gene and phenotype should be identified. Because lexical features are relatively regular, there are many methods to identify genes in the text. (Campos et al., 2012; Wei et al., 2015) However, although research on NER has been improved (Gaizauskas et al., 2003; Horn et al., 2004; Segura-Bedmar et al., 2008), phenotype identification is still challenging and this negatively influences relationship extraction.
First, a phenotype is usually composed of multiple words, such as ‘calcium sensitivity’ or ‘genic male sterility-photoperiod sensitive’. Thus, name boundaries are complex. Second, phenotypic descriptions are often study- or author-specific due to a lack of standard expressions, complicating this search. For example, in the two sentences ‘…resulting in root growth inhibition, smaller rosettes, and leaf curling'. (PMID: 26734017) and ‘…leading to early flowering and curly leaves phenotypes’. (PMID: 25693187), the same leaf morphology has two different descriptions, i.e. ‘leaf curling’, ‘curly leaves’. In addition, while there are specialized lexicons in many areas, no lexicon can be directly used to identify overall phenotypic descriptions in text, especially for plants. For example, the Unified Medical Language System (UMLS) MetaThesaurus (Humphreys et al., 1998) is a vocabulary database that includes numerous semantic types, except for Phenotype type. In the plant domain, the controlled vocabulary plant trait ontology (PTO) (http://bioportal.bioontology.org/ontologies/PTO ) is too general, so it may not include all species traits. The Arabidopsis Information Resource (TAIR) (Lamesch et al., 2012) is curated by manually summarizing published literature so it is limited and difficult to organize for future use. The AraPheno (Seren et al., 2017) database is an organization of the Genome-Wide Association Study (GWAS) phenotypic results in only six published studies, so the data are few. These manual curation processes are time-consuming and cannot keep up with rapidly increasing literature.
Here, we propose a novel gene–phenotype relationship extraction pipeline using model plant Arabidopsis thaliana. First we improved the word-embedding-to-sentence-embedding cascaded approach (Xing et al., 2017) as representation learning to recognize various broad phenotypic descriptions in large-scale biomolecular literature. Then, genes from the same phenotype-containing sentence were found, using the dictionary-based method. Next, a relationship extraction system Open Language Learning for Information Extraction (OLLIE) was applied to extract gene–phenotype relationships.
The proposed pipeline improves relationship extractions by identifying more phenotypic descriptions in the text. We identified many types of phenotypic descriptions based on their boundary delimitation: phenotypic phrases and phenotypic long/short sentences. To locate sentences that include the phenotype, we use word embedding to learn distributed representations for words and phrases. Then, we can extract phenotypic phrases missed by ontology, thus extracting more sentences containing phenotypes. Then we cascade the sentence-embedding method for specific phenotype-containing sentences. Due to numerous candidate phenotypic sentences, expert verification is time-consuming. According to the similarity mechanism, we find that sentences with high similarity to the phenotype-containing sentences have similar sentence structures. This prompted us to design a Phenotypic Sentence Template Extraction arChitecture (PSTEC) algorithm that automatically extracts phenotype sentence templates. With these templates, we can extract complex non-phrase forms of long/short phenotypic sentences.
Ultimately, we evaluated the proposed pipeline from two aspects. (i) We designed three baselines to compare with our proposed relationship extraction pipeline. From the results, we identified more phenotypes (expanding the original ontology almost 3-fold), which significantly improved recall value (improving 24.05% compared to the traditional ontology-based method). Meanwhile, identifying phenotypic descriptions from multiple perspectives also increased the precision of whole recognition. Using the OLLIE system based on machine learning method, we effectively improved F1-Measure compared with traditional relation extraction approach. Thus, our pipeline had a F1-Measure of 66.83%, the greatest of all baselines. (ii) We applied the pipeline to 481 full articles from the TAIR gene–phenotype relationship dataset, and the coverage was 70.94%. Moreover, we added 373 relationships to expand this dataset. Our pipeline automatically identified new relationships with a growing body of literature showing strong scalability. The proposed pipeline is versatile and can be used not only for extraction of relationships in Arabidopsis but also for other plant species such as soybean and cotton.
2 Our gene–phenotype relationship pipeline
2.1 The overview of our pipeline
The pipeline starts with scanning abstracts in PubMed using the keyword ‘A.thaliana’ and the Entrez Programming Utilities (E-utilities) web service (https://www.ncbi.nlm.nih.gov/books/NBK25501). We clean irrelevant author information and acquire 63 459 abstracts that mention A.thaliana.
Next, we improve the proposed cascaded representation learning approach (Xing et al., 2017) to recognize various broad phenotypes in the literature. Our representation learning approach, combined with the syntactic and semantic analysis of texts, identifies phenotypes in multiple directions from phenotypic phrases to complex short/long phenotypic sentences. Using ontology terms as input, our approach greatly expands the recognition of ontology term synonyms in the literature and establishes a bridge from ontology to literature description, so that study- or author-specific terms can be identified.
Then we use the results of phenotypic identification to extract gene–phenotype relationships. We use dictionary- and rule-based methods to identify Arabidopsis genes in the literature. Then, we combine the workflow of the Open Information Extraction (IE) system with our entity recognition to extract and establish an Arabidopsis gene–phenotype binary relationship. The pipeline was implemented and run on a 24 2.4 GHz Xeon core server running on Ubuntu Linux 16.04. Figure 1 shows the overview of the pipeline.
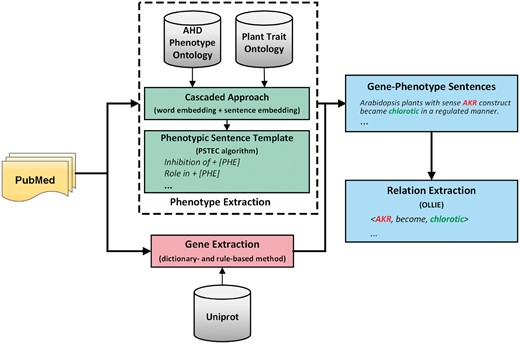
The overview of our gene–phenotype relationships extraction pipeline
2.2 Cascaded approach for phenotype extraction
Before entity recognition, we used domain-resource ontology to establish the original phenotypic dataset. We extracted phenotypic descriptions from phenotypic phrases and sentences based on different boundaries. We used the parse tree combined with the word embedding method to extract phenotypic phrases, the majority of which were described by noun phrases. Because some synonyms in ontology are not described as phenotype in the text, the previous approach did not consider it leading to some errors. Therefore, we added abbreviation recognition and revision algorithm into the improved cascaded approach.
Because some special phenotypes are non-phrase forms or long/short sentence descriptions, we used phenotypic sentences from word embedding results as positive samples to cascade the sentence embedding method for finding phenotype sentences. We transformed the unsupervised sentence-embedding model into a weakly supervised model. Due to the lack of training of positive and negative samples, we use the Negative Class Label Enhanced (NCLE) algorithm (Xing et al., 2017) to label negative samples and train the sentence-embedding model in combination with the positive samples of the word-embedding results. We analyzed results of sentence embedding, finding that phenotypic sentences gathered by the similarity mechanism had similar structures. However, the previous approach estimated these results through expert verification, which is time-consuming. Therefore, we extracted sentence templates that described the phenotype by improving the algorithm of the statistical combination to expand phenotype recognition.
2.2.1 Constructing the phenotype dataset
First, we use two ontologies to create the original phenotype dataset P, i.e. PTO and Arabidopsis Hormone Database 2.0 (http://ahd.cbi.pku.edu.cn/cgi-bin/phenotypeBrowse.pl .) (Jiang et al., 2011). PTO is an important controlled vocabulary that describes phenotypic traits in plants. Each trait is a distinguishable, characteristic, quality or phenotypic feature of a developing or mature plant or a plant part. Arabidopsis Hormone Database 2.0 provides a systematic and comprehensive view of genes participating in plant hormonal regulation of the model organism A. thaliana. Its phenotypic ontology was developed to describe precisely myriad hormone-regulated morphological processes with standardized vocabularies in Arabidopsis.
When processing PTO, we extract ‘name’ and ‘synonym’ from every term in the ontology. Approximately 84% of these names are associated with synonyms; on average, each name has 1.07 synonyms. For example, the phenotype ‘alkali soil sensitivity’ has two synonyms: ‘AlkS’ and ‘alkali sensitivity’. Not all of terms in these ontologies appear in the literature. We found 805 terms in abstracts after removing duplicate entries. We combined these into a complete phenotype dataset P.
2.2.2 Word embedding
We followed the word embedding method published in (Xing et al., 2017). First, we used the collected PubMed texts to train the word-embedding model, which gave each word or phrase a distributed representation in low and dense dimensional vector space. By finding phrases with high similarity to phenotypic entities in P, the original ontology of the phenotype is expanded as Pupdate. Therefore, we can obtain more sentences containing phenotypic information.
Because some phenotypic synonyms contained in P are abbreviated forms, they may not represent as phenotype in the text and are incorrectly identified. For example, the abbreviation ‘AC’ in the ontology corresponds to the full name of ‘leaf sheath auricle color’. However, in the sentence ‘Many of these proteins have complex domain architectures with AC or GC centers …’ (PMID: 26721677), ‘AC’ is not a phenotype. The previous method did not consider abbreviation recognition such as this, so we required post-processing of word-embedding results. After obtaining a high similarity phenotype phrase, we recognized and revised the abbreviation.
We used (Xu et al., 2009) algorithms for identifying abbreviations in the biological literature, matching pairs of all abbreviations and full names in the processed texts. When we used an updated phenotype dataset Pupdate to reidentify the phenotype in the literature, if there was an abbreviated form, it was first matched with a full name. Only the full name of the abbreviation also in Pupdate, remained as a phenotype, otherwise it was deleted. The abbreviation recognition and revision can increase pipeline precision value and identify phenotypes more accurately.
2.2.3 Sentence embedding
Using the word-embedding results, we classified and tagged PubMed texts as input for the sentence-embedding (Le and Mikolov, 2014) method. The trained model can find sentences containing phenotypic information, acquiring new phenotypic sentences. To improve diversity of phenotype recognition, we transformed the unsupervised sentence-embedding model into a weakly supervised model. We used the results of word-embedding as positive samples, Spos, and combined the NCLE algorithm for negative samples, Sneg, for the training of the Sen2Vec model.
Sentence embedding can aggregate similar phenotypic expressions. We found that large-scale gathered sentences have a similar sentence context structure. For example, the more similar sentences with ‘Solute import across the pollen plasma membrane, which occurs via proteinaceous transporters, is required to support pollen development and also for subsequent germination and pollen tube growth’ always have the same structure ‘be required {prep_*} + [phenotype]’, such as:
‘During pollination, constant communication between male pollen and the female stigma is required for pollen adhesion, germination, and tube growth’.
‘Two A.thaliana genes, QRT1 and QRT2, are required for pollen separation during normal development’.
Due to many similar sentences, it is time-consuming to identify all phenotypic sentences and analyze their phenotype with expert evaluation. Therefore, we used sentence structure to automate extraction of complicated long/short phenotypic sentences of non-phrase types. These structures may contain complex phenotypic descriptions, likely with punctuation, prepositions, and conjunctions. We designed an automated algorithm to find frequently occurring sentence templates and with this, we extracted relatively complex descriptions of phenotypic long/short sentences from many sentence-embedding results.
At present, there are few studies about automatic generation of sentence templates in NLP. We borrowed the idea of modular algorithms from Sentence Pattern Extraction arChitecturte (SPEC) systems in (Michal et al., 2011) and proposed our own solution for combinatorial explosion problem.
With the SPEC algorithm, a ‘sentence template’ is considered as n-element ordered combination of sentence elements. It generates all possible combinations of patterns from a sentence and selects the frequency occurrence combination as a sentence pattern. However, we focused on the phenotype-containing structure and created the algorithm Phenotypic Sentence Template Extraction arChitecture (PSTEC) which consists of three components:
Preprocessing
Generation of all ordered combinations from sentence elements
Insertion of a wildcard
Preprocessing: We tokenized all positive sentences Spos of sentence embedding. Because we must extract phenotype-containing sentence structures, we treated phenotypic phrases as a whole and replaced phenotypic descriptions appearing in the sentence with ‘PHE’.
Generation ordered combinations: In every n-element sentence, there is k-number of ordered combination groups (1 ≤ k ≤ max). After processing all sentences in corpora, we choose a combination of frequencies greater than a threshold fre as a k-length template. Because the phenotype-containing template is not too long, so we set max as the length of the element threshold. We set two restrictions to prevent the combination explosions:
Combination of the k-element must include the specific word ‘PHE’
Any ‘PHE’ contained (k-1)-element subset of k-element combination must be in the (k-1)-element template.
After iteration processing, we obtained all ordered, not duplicated, high frequency combinations for all values of k from the range of {1, …, max} as k-element sentence templates.
Insertion of a wildcard: During combination, we combined the original word order. To improve the applicability of templates, we specified whether the elements appeared next to each other or were separated. Therefore, we placed a wildcard between all non-subsequent elements using one heuristic rule. If an absolute difference of word order assigned to the two subsequent elements of a combination >1, we added a wildcard between them. An example of PSTEC algorithm appears in Figure 2.
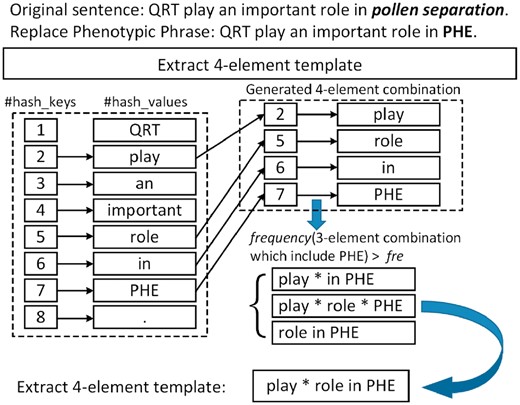
The procedure for sentence template extraction using high frequency three-element combinations to generate four-element template
When we obtained the high-frequency max-element sentence templates, we applied these templates to the results of a large number of sentence embeddings. Extracting the description of the more complex phenotypes in sentences that are highly similar to the positive samples improved phenotype recognition.
2.3 Gene–phenotype relationship extraction
For gene–phenotype relationship extraction, the gene is required and gene lexical features are relatively regular in texts, gene IDs or gene names may be used to represent them. Therefore, we used a dictionary- and rule-based method to identify genes.
After entity recognition was complete, our pipeline extracted the relationship with the open information extraction (IE) system. Results of the relationship extraction are expressed as triplets (arg1; r; arg2). The r (relationship phrase) represents arg1 and arg2 entity relationships.
2.3.1 Gene extraction
First, we searched all related genes in the UniProt database (http://www.uniprot.org ) using ‘A.thaliana’ as a key word and obtained 129 648 records. Each record contained the fields ‘Organism’, ‘Gene locus’, ‘Gene name’. Although we use Arabidopsis as a keyword, the results included other species, such as ‘Oryza sativa subsp. japonica (Rice)’. After processing, we obtained 89 287Arabidopsis gene ID and gene name pairs and these were used as a dictionary to identify genes.
Due to the large number of gene names and not a gene locus in the literature, part of the gene name is not in the dictionary. Therefore, we use gene lexical rules and semantic description rules in the text to improve gene recognition. Gene name spelling had some character-level rules as follows:
All capital letters.
A combination of uppercase and lowercase letters.
A combination of numbers, uppercase and lowercase letters.
Those containing hyphens.
Therefore, we used two rule types, mixed character-levels and contextual-levels, to identify the gene. When an input sentence contained these expressions: Expression of, Accumulation of, Expression levels/patterns of, Targets of, mRNA abundance of, Transcript profiles/levels of, and the ‘NNP’ (Proper noun, singular) tagged parts in the part-of-speech (POS) tagged sentence complies with our character-level rules, we extracted this special expression as a gene. For example, with the POS tagged sentence: “…HTR4K27Q (‘NNP’) overexpression (‘NN’) lines (‘NNS’) exhibited (‘VBD’) deregulated (‘JJ’) expression (‘NN’) of (‘IN’) H3K27me3-enriched (‘NNP’) genes (‘NNS’).” (PMID: 27926813) contains the specific contextual-level description ‘Expression of ’, and the ‘NNP’ tagged words satisfy the third and fourth character-level rules. Thus, we can identify gene ‘H3K27me3-enriched’.
Then, we used all sentences that contained the phenotype as input, and the output is two entities that cooccur in sentences. These sentences were used as input to subsequent relationship extraction.
2.3.2 Relationship extraction
To the best of our knowledge, there is a limited document annotation corpus of gene–phenotype relationships in Arabidopsis species. Currently we are only concerned with gene–phenotype relationships in single sentences. Most relationship recognition systems are not generic and portable so we used the open information extraction (OpenIE) system for this specific relationship identification. OpenIE can extract assertions from massive corpora without a specified vocabulary (Fader et al., 2011) from open-domain corpora, such as the Internet and Wikipedia, but in recent years, OpenIE has used biological literature for systematic testing.
We used an existing OpenIE system, OLLIE (Schmitz et al., 2012) as a relationship phrase recognition tool. OLLIE improved several shortcomings of the state-of-the-art system, extracting only relationships mediated by verbs and ignoring context, extracting tuples not asserted as factual. OLLIE is popular for information extraction and used in many fields, such as Question-Answer (Berant et al., 2013), knowledge graphs (Nickel et al., 2016), and named entities’ network (Tariq et al., 2017). OLLIE uses high-precision results of the previous generation OpenIE system i.e. REVERB (Fader et al., 2011). With many syntactic analyses of sentences that contain relationships, learning relationship patterns can be extended to find relationships of new input sentences.
We input the co-occurring sentences into the OLLIE system and extracted relationship sentences and their corresponding relationships. OLLIE automatically gives NP pairs of sentences as arguments in the relationship. However, these NP pairs contain too much noise, and the partially extracted arguments are not genes or phenotypes. Therefore, we limited our screening to eligible relationship groups. For the first (agr1) and third (agr2) parts of one triple, we need map them to the previous phenotype and gene entity list. When one or some genes and phenotypes are in each of the two arguments, we consider the relationship as a gene–phenotype relationship and stored such a relationship.
3 Results and discussion
3.1 Phenotype extraction results
3.1.1 Word-embedding results
We used Word2Vec (https://code.google.com/p/word2vec ) to train a skip-gram model with a 4 D size, i.e. 300, 500, 700 and 900. Due to a lack of standards for this topic, we needed expert evaluation and annotation. Therefore, the results of word embedding first were semi-automatically classified and then manually evaluated by one expert and confirmed by another. Ultimately, the word-embedding method can extend original phenotype datasets P, increasing 1303 new phenotype data by up to 161.86%. We used the extended dataset Pupdate to match the phenotypic descriptions in the abstracts. Mapping sentences numbered 88 243. After abbreviations were identified and revised, 87 613 sentences containing phenotypes were obtained.
Some examples of phenotypes recognized by the word-embedding method appear in Table 1. ‘Ontology term’ as the original input, using the similarity mechanism to get ‘Phenotype’ and the corresponding ‘Similarity Score’. ‘Class’ represents the corresponding categories in the PTO 10 basic categories (10 basic categories are: TO: 0000277 biochemical trait; TO: 0000283 biological process trait; TO: 0000183 other miscellaneous trait; TO: 0000357 plant growth and development trait; TO: 0000017 plant morphology trait; TO: 0000597 quality trait; TO: 0000133 stature or vigor trait; TO: 0000392 sterility or fertility trait; TO: 0000164 stress trait; TO: 0000371 yield trait).
Examples of word-embedding results
| Ontology term | Phenotype | Similarity Score | Class |
|---|---|---|---|
| Cell elongation | Cell expansion | 0.671 | TO: 0000357 |
| Cell enlargement | 0.531 | ||
| Organ expansion | 0.528 | ||
| Cell proliferation | 0.526 | ||
| Chlorophyll content | Lower ion leakage | 0.625 | TO: 0000277 |
| Photosystem II activity | 0.557 | ||
| Photosynthetic quantum yield | 0.550 | ||
| Higher relative water content | 0.531 | ||
| Chloroplast structure | Photosynthetic phenotype | 0.498 | TO: 0000017 |
| Thylakoid structure | 0.495 | ||
| Leaf chloroplast ultrastructure | 0.484 | ||
| Pale green leaves | 0.479 | ||
| Leaf curling | Dark green leaves | 0.613 | TO: 0000357 |
| Altered leaf shape | 0.581 | ||
| Curly leaves | 0.576 | ||
| Serrated leaves | 0.558 | ||
| Drought sensitivity | Reduced water loss | 0.550 | TO: 0000164 |
| Enhanced drought resistance | 0.544 | ||
| Drought stress tolerance | 0.539 | ||
| Reduced drought tolerance | 0.535 |
| Ontology term | Phenotype | Similarity Score | Class |
|---|---|---|---|
| Cell elongation | Cell expansion | 0.671 | TO: 0000357 |
| Cell enlargement | 0.531 | ||
| Organ expansion | 0.528 | ||
| Cell proliferation | 0.526 | ||
| Chlorophyll content | Lower ion leakage | 0.625 | TO: 0000277 |
| Photosystem II activity | 0.557 | ||
| Photosynthetic quantum yield | 0.550 | ||
| Higher relative water content | 0.531 | ||
| Chloroplast structure | Photosynthetic phenotype | 0.498 | TO: 0000017 |
| Thylakoid structure | 0.495 | ||
| Leaf chloroplast ultrastructure | 0.484 | ||
| Pale green leaves | 0.479 | ||
| Leaf curling | Dark green leaves | 0.613 | TO: 0000357 |
| Altered leaf shape | 0.581 | ||
| Curly leaves | 0.576 | ||
| Serrated leaves | 0.558 | ||
| Drought sensitivity | Reduced water loss | 0.550 | TO: 0000164 |
| Enhanced drought resistance | 0.544 | ||
| Drought stress tolerance | 0.539 | ||
| Reduced drought tolerance | 0.535 |
Note: According to the original ‘Ontology term’, we use similarity mechanisms to extract ‘Phenotype’ and its corresponding ‘Similarity Score’. ‘Class’ represents the corresponding categories in the PTO 10 basic categories.
Examples of word-embedding results
| Ontology term | Phenotype | Similarity Score | Class |
|---|---|---|---|
| Cell elongation | Cell expansion | 0.671 | TO: 0000357 |
| Cell enlargement | 0.531 | ||
| Organ expansion | 0.528 | ||
| Cell proliferation | 0.526 | ||
| Chlorophyll content | Lower ion leakage | 0.625 | TO: 0000277 |
| Photosystem II activity | 0.557 | ||
| Photosynthetic quantum yield | 0.550 | ||
| Higher relative water content | 0.531 | ||
| Chloroplast structure | Photosynthetic phenotype | 0.498 | TO: 0000017 |
| Thylakoid structure | 0.495 | ||
| Leaf chloroplast ultrastructure | 0.484 | ||
| Pale green leaves | 0.479 | ||
| Leaf curling | Dark green leaves | 0.613 | TO: 0000357 |
| Altered leaf shape | 0.581 | ||
| Curly leaves | 0.576 | ||
| Serrated leaves | 0.558 | ||
| Drought sensitivity | Reduced water loss | 0.550 | TO: 0000164 |
| Enhanced drought resistance | 0.544 | ||
| Drought stress tolerance | 0.539 | ||
| Reduced drought tolerance | 0.535 |
| Ontology term | Phenotype | Similarity Score | Class |
|---|---|---|---|
| Cell elongation | Cell expansion | 0.671 | TO: 0000357 |
| Cell enlargement | 0.531 | ||
| Organ expansion | 0.528 | ||
| Cell proliferation | 0.526 | ||
| Chlorophyll content | Lower ion leakage | 0.625 | TO: 0000277 |
| Photosystem II activity | 0.557 | ||
| Photosynthetic quantum yield | 0.550 | ||
| Higher relative water content | 0.531 | ||
| Chloroplast structure | Photosynthetic phenotype | 0.498 | TO: 0000017 |
| Thylakoid structure | 0.495 | ||
| Leaf chloroplast ultrastructure | 0.484 | ||
| Pale green leaves | 0.479 | ||
| Leaf curling | Dark green leaves | 0.613 | TO: 0000357 |
| Altered leaf shape | 0.581 | ||
| Curly leaves | 0.576 | ||
| Serrated leaves | 0.558 | ||
| Drought sensitivity | Reduced water loss | 0.550 | TO: 0000164 |
| Enhanced drought resistance | 0.544 | ||
| Drought stress tolerance | 0.539 | ||
| Reduced drought tolerance | 0.535 |
Note: According to the original ‘Ontology term’, we use similarity mechanisms to extract ‘Phenotype’ and its corresponding ‘Similarity Score’. ‘Class’ represents the corresponding categories in the PTO 10 basic categories.
As shown in Table 1, the word-embedding method can find a phenotypic description according to the syntax and context of the text. For example, for the same ontology term ‘leaf curling’ (TO: 0002681), the method can extract similar words by considering syntax (‘leaf curling’—‘curly leaves’) and context semantics (‘leaf curling’—‘altered leaf shape’). Some new phenotypes are not synonyms of their corresponding original ontology terms. For example, the new phenotype ‘serrated leaves’ is not synonymous with ‘leaf curling’. This may because the contextual environment that describes the new phenotype and the original term is similar, but the semantics of expression are not the same.
3.1.2 Sentence-embedding results
We used Doc2Vec (http://radimrehurek.com/gensim/models/doc2vec.html ) to train the PV-DBOW model, and the trained corpora are positive/negative labeled abstracts. Then, we used the results of word embedding Spos as inputs and acquired candidate sentences with similarities greater than Sim after calculating for cosine distance with Spub. A reasonable Sim value greatly influenced the results. After testing, if Sim was too high (>0.4), high similarity sentences were too few and an average of 1.2 high-similarity sentences was obtained for each original sentence. If Sim was too low (¡0.2), we get a lot of dissimilar sentences. Therefore, we set Sim as 0.3, and an average of 4.5 high-similarity sentences was obtained for each original sentence.
The sentence-embedding method can find many candidate phenotypic sentences, which contain many non-phrase, complex long/short phenotypic sentences. For example, the phenotypic structure ‘response to …stress’ in the sentence ‘GmaPHO1 genes had altered expression in response to salt, osmotic, and inorganic phosphate stresses’. Such phenotypic descriptions are special and numerous and can improve relationship identification. Therefore, we designed a PSTEC algorithm to automatically generate phenotypic sentence templates for extracting them.
We tested and selected template length max and template frequency fre of the PSTEC algorithm. When max is too long (>6), the template will contain a lot of noise, such as too many prepositions and stop words. When max is too short (<4), the template cannot contain complete template structure information. Thus, we set max as 5. The size of fre directly affects the efficiency and uptime of the algorithm. After testing, we set fre as 100 and only kept templates that appeared more often than 100 in the corpus. Ultimately, we obtained 250 sentence templates. There are many types of duplicate templates and high frequency but not intention-containing templates, such as ‘Show/Suggest + prep_*’. Therefore, we merged and selected these results. Table 2 shows 5 high frequency sentence templates that can recognize combination type phenotypes (‘Tolerance to salt/drought/methyl viologen stress in Arabidopsis’), with environmental or time factors (‘Hypocotyl growth in response to unilateral blue-light illumination’) and are rich in diversity of phenotypes. Meanwhile, we noticed that phenotypes recognized by different templates may differ. For example, the ‘Respond’ template can identify more ‘stress trait’ types.
Examples of sentence templates
| Sentence template | Example of phenotype | Number of phenotype |
|---|---|---|
| Inhibition of + (PHE) | Root growth the root-swelling phenotype; Germination and elongation of Arabidopsis seedling | 127 |
| Involve(d) in + (PHE) | Host cell death in the hypersensitive disease-resistance response; A. thaliana seedling root to a rapid change in salinity | 532 |
| (Play a/an adj./n.) Role in + (PHE) | Coordinate the directional growth of plant tissue; Tolerance to salt/drought/methyl viologen stress in Arabidopsis | 243 |
| Regulator/regulation of + (PHE) | Secondary wall synthesis in fiber of A.thaliana stem; Stomatal clustering and density early in Arabidopsis leaf development | 197 |
| (In) Response to + (PHE) | Both high- and low-temperature stress; Signal emanate from cell undergo pathogen-induced hypersensitive cell death | 215 |
| Sentence template | Example of phenotype | Number of phenotype |
|---|---|---|
| Inhibition of + (PHE) | Root growth the root-swelling phenotype; Germination and elongation of Arabidopsis seedling | 127 |
| Involve(d) in + (PHE) | Host cell death in the hypersensitive disease-resistance response; A. thaliana seedling root to a rapid change in salinity | 532 |
| (Play a/an adj./n.) Role in + (PHE) | Coordinate the directional growth of plant tissue; Tolerance to salt/drought/methyl viologen stress in Arabidopsis | 243 |
| Regulator/regulation of + (PHE) | Secondary wall synthesis in fiber of A.thaliana stem; Stomatal clustering and density early in Arabidopsis leaf development | 197 |
| (In) Response to + (PHE) | Both high- and low-temperature stress; Signal emanate from cell undergo pathogen-induced hypersensitive cell death | 215 |
Note: PHE represents phenotype, parentheses indicate optional parts.
Examples of sentence templates
| Sentence template | Example of phenotype | Number of phenotype |
|---|---|---|
| Inhibition of + (PHE) | Root growth the root-swelling phenotype; Germination and elongation of Arabidopsis seedling | 127 |
| Involve(d) in + (PHE) | Host cell death in the hypersensitive disease-resistance response; A. thaliana seedling root to a rapid change in salinity | 532 |
| (Play a/an adj./n.) Role in + (PHE) | Coordinate the directional growth of plant tissue; Tolerance to salt/drought/methyl viologen stress in Arabidopsis | 243 |
| Regulator/regulation of + (PHE) | Secondary wall synthesis in fiber of A.thaliana stem; Stomatal clustering and density early in Arabidopsis leaf development | 197 |
| (In) Response to + (PHE) | Both high- and low-temperature stress; Signal emanate from cell undergo pathogen-induced hypersensitive cell death | 215 |
| Sentence template | Example of phenotype | Number of phenotype |
|---|---|---|
| Inhibition of + (PHE) | Root growth the root-swelling phenotype; Germination and elongation of Arabidopsis seedling | 127 |
| Involve(d) in + (PHE) | Host cell death in the hypersensitive disease-resistance response; A. thaliana seedling root to a rapid change in salinity | 532 |
| (Play a/an adj./n.) Role in + (PHE) | Coordinate the directional growth of plant tissue; Tolerance to salt/drought/methyl viologen stress in Arabidopsis | 243 |
| Regulator/regulation of + (PHE) | Secondary wall synthesis in fiber of A.thaliana stem; Stomatal clustering and density early in Arabidopsis leaf development | 197 |
| (In) Response to + (PHE) | Both high- and low-temperature stress; Signal emanate from cell undergo pathogen-induced hypersensitive cell death | 215 |
Note: PHE represents phenotype, parentheses indicate optional parts.
We can extend 1314 phenotypic descriptions using the sentence template. After merged results of word-embedding, we expanded 2409 phenotypic expressions and increased them 2.99-fold compared to the original phenotype dataset P.
3.2 Gene–phenotype relationship results
We evaluated results of gene–phenotype extraction from two perspectives.
According to different phenotype recognition and relation extraction methods, we compared with baselines.
We used the entire pipeline in the TAIR database, which manually extracted gene–phenotype relationships from 555 full papers.
3.2.1 Performance comparison with baselines
Using the phenotype recognition cascaded approach can improve the identification of phenotypes in the literature and improve relationship identification. To illustrate the importance of phenotypic recognition in relationship extraction and to verify the accuracy of our approach, we establish two baselines for performance comparison.
B1: Using the traditional ontology-based method (Müller et al., 2004) to recognize phenotype and extracting the gene and relationship using method described in this article.
B2: Using the ontology-based with word embedding method (Mikolov et al., 2013) to recognize phenotype and extracting the gene and relationship using method described in this article. we also compare with another baseline that use traditional relation extraction methods.
B3: Using method described in this article to extract phenotype and gene, the relation extraction method is based on syntatic rules (Coulet et al., 2010) which uses the collapsed dependencies graph representation.
We randomly selected 100 abstracts to identify the relationships by expert verification and to calculate Precision, Recall, F1-Measure. Results are shown in Table 3.
Performance of baselines compared with our pipeline
| Type | Phenotype extraction | Relation extraction | Precision (%) | Recall (%) | F1-Measure (%) |
|---|---|---|---|---|---|
| B1 | Ontology-based (Müller et al., 2004) | OLLIE | 52.98 | 33.76 | 41.24 |
| B2 | Ontology-based + word embedding (Mikolov et al., 2013) | OLLIE | 73.91 | 50.21 | 59.80 |
| B3 | Representation learning approach | Syntatic rules (Coulet et al., 2010) | 55.75 | 26.58 | 36.00 |
| Our pipeline | Representation learning approach | OLLIE | 79.19 | 57.81 | 66.83 |
| Type | Phenotype extraction | Relation extraction | Precision (%) | Recall (%) | F1-Measure (%) |
|---|---|---|---|---|---|
| B1 | Ontology-based (Müller et al., 2004) | OLLIE | 52.98 | 33.76 | 41.24 |
| B2 | Ontology-based + word embedding (Mikolov et al., 2013) | OLLIE | 73.91 | 50.21 | 59.80 |
| B3 | Representation learning approach | Syntatic rules (Coulet et al., 2010) | 55.75 | 26.58 | 36.00 |
| Our pipeline | Representation learning approach | OLLIE | 79.19 | 57.81 | 66.83 |
Performance of baselines compared with our pipeline
| Type | Phenotype extraction | Relation extraction | Precision (%) | Recall (%) | F1-Measure (%) |
|---|---|---|---|---|---|
| B1 | Ontology-based (Müller et al., 2004) | OLLIE | 52.98 | 33.76 | 41.24 |
| B2 | Ontology-based + word embedding (Mikolov et al., 2013) | OLLIE | 73.91 | 50.21 | 59.80 |
| B3 | Representation learning approach | Syntatic rules (Coulet et al., 2010) | 55.75 | 26.58 | 36.00 |
| Our pipeline | Representation learning approach | OLLIE | 79.19 | 57.81 | 66.83 |
| Type | Phenotype extraction | Relation extraction | Precision (%) | Recall (%) | F1-Measure (%) |
|---|---|---|---|---|---|
| B1 | Ontology-based (Müller et al., 2004) | OLLIE | 52.98 | 33.76 | 41.24 |
| B2 | Ontology-based + word embedding (Mikolov et al., 2013) | OLLIE | 73.91 | 50.21 | 59.80 |
| B3 | Representation learning approach | Syntatic rules (Coulet et al., 2010) | 55.75 | 26.58 | 36.00 |
| Our pipeline | Representation learning approach | OLLIE | 79.19 | 57.81 | 66.83 |
Among the different methods on phenotype recognition, the effect of recognizing the gene–phenotype relationship using only ontology-based efforts is the poorest. Because of loss of many phenotypes, recall value in relationship recognition is low. For example, the phenotype ‘NaCl stress-sensitive phenotype’ is not in ontology, so the relationship (MCK1; complemented; NaCl stress-sensitive phenotype) cannot be found. However, we can identify this phenotype using the proposed approach and obtain relationships with the best recall. This is because we recognized the phenotypic phrase and the more complex phenotypic long/short sentences based on the sentence template. As the integrity of the phenotype increased, the precision is improved. For this sentence, ‘…a structurally related Arabidopsis MADS-box gene involved in the negative control of Arabidopsis flowering time, …’ (PMID: 15539492), due to the template: ‘(gene) involve + {prep.} + PHE’, we can identify the whole phenotypic description ‘negative control of Arabidopsis flowering time’, and get the relationship (MADS-box gene; involved in; negative control of Arabidopsis flowering time). However, the first two baselines only extracted part of the whole expression ‘flowering time’ and missed the complete relationships. Thus, our approach can extend relationship extraction by improving phenotype recognition.
Compared with the B3 baselines, which only change the relation extraction method, our pipleline also has the best performance. Because the syntactic rule method misses many results and only getting 26.58% recall value, its F1-Measure is about 36.00%.
We have considered to use generic tools such as GNormPlus (Wei et al., 2015), GenNorm (Wei and Kao, 2011) and so on for gene identification but found that these tools identify the gene of all species that appear in the text. Therefore, noise information is mixed in the targeted identification of Arabidopsis gene information, which requires expert screening. So, we finally chose a more targeted rule- and dictionary-based approach and obtained 88.76% precision value in the above test dataset. This is slightly higher than the results given in the article (Wei et al., 2015) by GNormPlus (precision 87.1%) and GenNorm (precision 78.9%).
Although the proposed pipeline can improve the effectiveness of final relationship identification compared with baselines, there are misidentifications and omissions due to the following reasons:
Error of relationship recognition. The OLLIE system is limited as it can only identify the relationship in a single sentence, and the length of the sentence cannot be too long. Sentences >20 words have increased errors for relationship analysis (Schmitz et al., 2012). For example, the sentence ‘Hence, the narrow organ shape, reduced plant height, and reduced whorl 4 organ primordia are consistent with a general reduction of cell number, and, perhaps, reflect a role of SEU in promoting cell proliferation’ can be assessed by OLLIE to get this relationship (whorl 4 organ primordia; perhaps reflect; a role SEU in promoting cell proliferation). The wrong relationship association results in inaccurate identification of it.
Inaccurate phenotypic boundary. Although we can identify phenotype from phrases and long/short sentences, more complex phenotypes cause errors or incomplete identification. For example ‘The AGAMOUS gene of Arabidopsis is necessary for the proper development of stamens and carpels and the prevention of indeterminate growth of the floral meristem’. We did not recognize this sentence structure, resulting in incomplete recognition of relationships.
Problem of gene recognition. Although we use a relatively complete Arabidopsis gene database as a dictionary for gene ID and gene name identification, and get high precition value of 88.76%, the database may still missing some gene name as well as the corresponding relationship for it.
These errors reduce precision and recall because each case results in an incorrect or incomplete relationship extraction.
3.2.2 Comparison with TAIR
The TAIR database (Lamesch et al., 2012) is one of the most informative databases for storing Arabidopsis information, which contains a gene–phenotype relationships dataset. This information was manually extracted from 555 full texts. To verify pipeline effectiveness, we calculated coverage of relationship for these papers. Because some documents cannot be downloaded, we retrieved only 481 full papers. Preprocessing the TAIR dataset by deleting irrelevant fields, i.e. ‘Phenotype not described’ and ‘No visible phenotype’ was done and we retrieved 1397 sets of gene–phenotype relationships. We noticed that there are duplicate types of relationships in the dataset. For example, the gene ‘MSSP1’ is related with:
Under normal growth temperature conditions, the double mutant leaves’ content in glucose and fructose is slightly reduced (30%) in a similar fashion to that observed with the tmt1 single mutants.
Under normal temperature conditions, a substantial reduction in glucose and fructose contents in leaves is observed compared to wild type, and even the single tmt1 and double tmt1/tmt2 mutants.
As they are the same type, we treat them as the identical relationships. We applied our pipeline to this dataset, extracted data were compared with processed TAIR datasets by four experts and offered coverage of 70.94%. Moreover, our pipeline can identify 373 new relationships, which the TAIR dataset does not include. The results are shown in Supplementary Material. We had limited coverage for a few reasons:
Many relationships in TAIR come from cross-sentence or even cross-paragraph relationships. Such relationships are unrecognizable to our pipeline that only extracts from a single sentence, so there is the main reason of limited coverage. However, due to redundancy of much information, our pipeline use repetitive relationships extracted from many studies to compensate extraction of the relationship representations in small samples. Such work cannot be done manually.
Many phenotypes in TAIR have not been described in the original literature after subsequent manual processing and summary and this will influence coverage.
There is only a gene locus name in the TAIR dataset, but most documents only describe the gene name. Some gene loci in the gene database do not have corresponding names. Thus, our pipeline cannot recognize these genes or any corresponding relationships.
After analysis, we found that articles in the TAIR dataset are relatively old (most prior to 2000). Due to limitations to manual reading, this dataset failed to update gene–phenotype relationships as the literature grew, so scalability was poor. However, with our pipeline we can quickly find relationships for updated literature, greatly improving efficiency for summarizing data.
4 Conclusion and future works
Much plant gene–phenotypic information exists in the biomedical literature, and it continues to grow. Thus, we propose a pipeline to extract relationships between genes and phenotypes using A.thaliana as an experimental object. Our pipeline can expand the expression of original phenotype ontology terms in the literature using an improved cascaded representation learning approach of phenotype recognition. This can enhance relationship extraction. Our pipeline obtained an F1-score (66.83%) that outperformed other baselines. Applying the pipeline to the TAIR dataset, we can complement 373 new relationships.
Future studies may include considering environmental influences and phenotypic conditions for constructing gene–phenotype event extraction instead of binary relationships. If the division of phenotype and relationship boundaries is more detailed, performance will be improved.
Funding
This work was supported by National Key Research and Development Program [grant no. 2016YFD0101900], National Natural Science Foundation of China [grant no. 31701144].
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Wenhui Xing and Junsheng Qi authors should be regarded as Joint First Authors.


{kind=link}
{kind=link}