





Abstract
Gene Ontology (GO) has been widely used to annotate functions of proteins and understand their biological roles. Currently only of >70 million proteins in UniProtKB have experimental GO annotations, implying the strong necessity of automated function prediction (AFP) of proteins, where AFP is a hard multilabel classification problem due to one protein with a diverse number of GO terms. Most of these proteins have only sequences as input information, indicating the importance of sequence-based AFP (SAFP: sequences are the only input). Furthermore, homology-based SAFP tools are competitive in AFP competitions, while they do not necessarily work well for so-called difficult proteins, which have sequence identity to proteins with annotations already. Thus, the vital and challenging problem now is how to develop a method for SAFP, particularly for difficult proteins.
The key of this method is to extract not only homology information but also diverse, deep-rooted information/evidence from sequence inputs and integrate them into a predictor in a both effective and efficient manner. We propose GOLabeler, which integrates five component classifiers, trained from different features, including GO term frequency, sequence alignment, amino acid trigram, domains and motifs, and biophysical properties, etc., in the framework of learning to rank (LTR), a paradigm of machine learning, especially powerful for multilabel classification.
The empirical results obtained by examining GOLabeler extensively and thoroughly by using large-scale datasets revealed numerous favorable aspects of GOLabeler, including significant performance advantage over state-of-the-art AFP methods.
Supplementary data are available at Bioinformatics online.
1 Introduction
The Gene Ontology (GO) was originally launched in 1998 for the consistent descriptions of gene and gene product (such as protein and RNA) across all species (Ashburner et al., 2000). Currently GO has >40 000 biological concepts over three domains: Molecular Function Ontology (MFO), Biological Process Ontology (BPO) and Cellular Component Ontology (CCO). Annotating protein function by GO is crucial and useful for understanding the nature of biology. With the development of next generation sequencing technology, we have seen the explosive increase of protein sequences, while the number of proteins with experimental GO annotations is limited, due to the high time and financial cost of biochemical experiments. In fact, only <1% of 70 million protein sequences in UniProtKB (The UniProt Consortium, 2015) have experimental GO annotations. To reduce this huge gap, an imperative issue would be efficient automated function prediction (AFP) (Jiang et al., 2016; Radivojac et al., 2013).
Automated function prediction is a large-scale multilabel classification problem (Zhang and Zhou, 2014) by regarding one GO term as a class label and also one protein as an instance with multiple labels (multiple GO terms). AFP is very challenging due to: (i) structured ontology: GO terms are node labels of a directed acyclic graph (DAG), by which one gene annotated at one node must be labeled by GO terms of all ancestor nodes in the DAG; (ii) many labels per protein: we checked GO terms in Swissprot (Boutet et al., 2016) (October 2016) with 66 841 proteins and found that a human protein is labeled by around 71 GO terms on average; (iii) large variation in the number of GO terms per protein: also we found that only 634, out of all 10 236 GO terms in MFO (GO Ontology in June 2016), are associated with >50 proteins. This means that most of GO terms are associated with only a small number of proteins.
To advance the performance of AFP, the first and second Critical Assessment of Functional Annotation (CAFA1 and CAFA2) challenges (Also currently (2016–2017), CAFA3 is on-going.)(competitions) were held in 2010–2011 and 2013–2014, respectively (Jiang et al., 2016; Radivojac et al., 2013). CAFA uses proteins that have no experimental annotations for each domain (i.e. MFO, BPO or CCO), before the prediction submission deadline. A target protein is called a limited-knowledge protein, if it has experimental annotation in another domain; otherwise, it is called a no-knowledge protein (Jiang et al., 2016). Importantly, this means no-knowledge proteins have only sequences and no experimental annotations for all domains before the deadline. Obviously in practice majority (around 99% or more) of proteins have only sequences, meaning that AFP for no-knowledge proteins would be more important. In fact experimental information, such as protein–protein interactions, are more costly than sequencing, resulting in literally limited knowledge (vastly missing information) among the large-scale data of AFP. Compared with limited experimental data, sequences can be the primary data available for various species. These would be also the reason why sequence-based AFP is important.
The results of CAFA for no-knowledge benchmark have shown that simple homology-based methods with BLAST and PSI-BLAST are very competitive (Altschul et al., 1997; Gillis and Pavlidis, 2013; Hamp et al., 2013). For example, Hamp et al. (2013) found that their implementations using simple homology-based inference was only slightly worse than the best method from the Jones group in CAFA1. This indicates that sequence identity is even at the present time still a key to achieve high performance of large-scale sequence-based AFP, and also implies that prediction would be hard for sequences of low identities with any other sequences. For example, the no-knowledge benchmark can be divided into two types, according to the largest global sequence identity of the corresponding sequence to any other sequences in the training data (Jiang et al., 2016). That is, one type, called difficult, includes those with the largest sequence identity of <60%; otherwise they are called easy. So an urgent issue for AFP is, instead of relying on sequence homology only, to develop an approach which can predict the function of the difficult type of proteins within the scope of sequence-based approach. An important point of this approach would be to collect not only homology-related information but also various types of diverse and informative sequence information and develop a method which can integrate all of these information effectively and also efficiently.
We propose a new method which we call GOLabeler for predicting functions of no-knowledge proteins, particularly for those in the difficult type. The basic idea of GOLabeler is to integrate different types of sequence-based evidence in the framework of ‘learning to rank’ (LTR; See Supplement for more introduction on LTR; Li, 2011). The idea of LTR is that for example, positive examples which are ranked lower are more penalized, while they are treated rather equally in regular classification. LTR was originally developed for ranking web pages to be consistent with the relevance between web pages and user queries. If we focus on binary relevance, the ranking problem turns into the problem of predicting relevant web pages for given queries. This is exactly multilabel classification, by regarding web pages as labels and queries as examples. LTR can solve this kind of problem by ranking labels and choosing top of them. So LTR can be applied to AFP by thinking GO terms as labels and proteins as examples. Another noteworthy advantage of LTR is that GOLabeler can integrate multiple sequence-based evidence effectively, which are generated by different types of classifiers (or components), where all information are derived from the sequences only.
We examined the performance of GOLabeler extensively by using large-scale datasets, which were generated by following the idea of using time-delayed performance evaluation procedure in CAFA. Particularly we compared the performance of GOLabeler with all component methods, three ensemble approaches and two sequence-based methods. The computational experimental results indicate significant performance advantage of GOLabeler over all competing methods in all experimental settings. In particular, the advantage of GOLabeler could be seen in the prediction for difficult proteins. Finally we present a typical result, showing that GOLabeler could predict the largest number of all GO terms correctly among all competing methods. Moreover, according to the initial evaluation results of CAFA3 (http://biofunctionprediction.org/meetings/), GOLabeler achieved the first place out of nearly 200 submissions from around 50 labs all over the world in terms of Fmax in all three GO domains.
2 Related work
A lot of biological information, such as protein structures, protein-protein interactions (Ma et al., 2014) and gene expression (Walker et al., 1999), are useful for AFP, while majority of proteins have no such information except for sequences (Shehu et al., 2016). We thus focus on sequence-based approaches, in which sequences or their parts are used in various ways, such as (i) sequence alignment, (ii) domains and motifs and (iii) features, etc., as follows: (i) sequence alignment: BLAST and/or PSI-BLAST are used to find homologous sequences and transfer their functional annotations to the query protein. For example, GoFDR (Gong et al., 2016), a top method in CAFA2, uses multiple sequence alignment (MSA) to generate position-specific scoring matrix (PSSM) for each GO term, to score the query against the corresponding GO term. (ii) domains and motifs: they are usually functional sites of a query protein, and all of them in the query sequence are detected by using protein domain/motif resources, such as CATH (Das et al., 2015), SCOP (Murzin et al., 1995) and Pfam (Sonnhammer et al., 1997), to understand the function of the query protein. (iii) Features: the protein query is an amino acid sequence, from which biophysical and biochemical attributes can be generated, which can be closely related with domains, motifs and protein families but not necessarily the same. ProFET (Protein Feature Engineering Toolkit) (Ofer and Linial, 2015) is a typical tool for extracting hundreds of such sequence-derived features including elementary biophysical ones. We note that these various sequence-based approaches play different roles, being complement to each other for AFP.
Thus integrating different types of information or classifiers trained from them would be a key to improve the performance of AFP. In fact in AFP, several approaches of using the idea of integrating data/classifiers have been already proposed. MS-kNN, a top method in CAFA1 and CAFA2, predicts the function by averaging over the prediction scores from three data sources, sequences, expression and protein-protein interaction (Lan et al., 2013) (Note that MS-kNN is NOT a sequence-based method). Also Jones-UCL, the top team of CAFA1, integrates prediction scores from multiple methods by using the ‘consensus’ function [given in Equation (1)] (Cozzetto et al., 2013). Recently different data integration methods, mainly ‘one vote’, ‘weighted voting’ and ‘consensus’, where ‘one vote’ relies on the classifier with the maximum confidence only, while ‘weighted voting’ weights over input classifiers (Khan et al., 2015; Lan et al., 2013).
All such data integration methods are rather simple integration technique, and this might underestimate the performance of integration. Our proposed approach, GOLabeler, is based on Learning to Rank (LTR), a powerful paradigm in machine learning for integrating multiple classifiers trained from different sequence-derived data. Recently LTR has been effectively used in bioinformatics, such as annotating biomedical documents (Liu et al., 2015; Peng et al., 2016) and predicting drug-target interactions (Yuan et al., 2016). LTR integrates the prediction results of component classifiers so that GO terms with being relevant more to the query protein should be ranked higher. One advantage of LTR is that the prediction results of components can be simply encoded as the input features of the model, over which any cutting edge classification/regression algorithm can be run. Thus, GOLabeler provides a nice framework of integrating different sequence-based information of AFP, being promising to improve the performance of the current AFP of no-knowledge proteins.
3 Materials and methods
3.1 Notation
Let D be the given training data with ND proteins, i.e. . Let Gi be the i-th GO term, and be the number of proteins with Gi in D (Note that this number is obtained by considering the structure of GO. That is, if Gi is assigned to a protein, this protein is with all GO terms of the ancestors of Gi in GO). Let T be the given test data (the number of proteins: ), in which let Pj be the j-th protein. Let be a binary indicator, showing if p is with ground-truth (true) Gi. That is, if p has ground-truth (true) Gi, is one; otherwise zero. Let be the score (obtained by a method), showing that Pj is with Gi. In particular in ensemble methods, is the predicted score between Gi and Pj by the k-th method (component).
3.2 Overview
Figure 1 shows the entire scheme of GOLabeler for AFP. In testing, given the sequence of a query protein, candidate GO terms are generated from five components, which are already trained by using different types of information. Each candidate GO term receives prediction scores from the five components, resulting in a feature vector of length five. Then candidate GO terms, i.e. feature vectors, are put into the learning to rank (LTR) model, which is also already trained by using training data, and finally, a ranked list of GO terms is returned as the final output of GOLabeler.
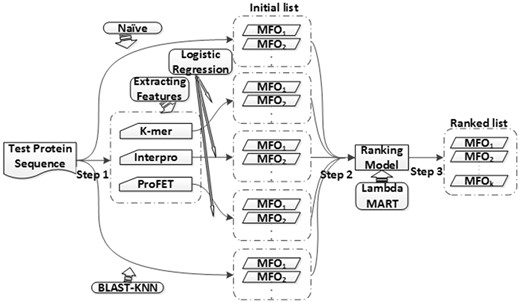
Entire scheme of GOLabeler with three steps for AFP
3.3 Component methods
We selected five typical, different sequence-based information for generating components. They are called Naive (GO term frequency), BLAST-KNN (B-K, k-nearest neighbor using BLAST results), LR-3mer [Logistic regression (LR) of the frequency of amino acid trigram], LR-InterPro (LR of InterPro features), and LR-ProFET (LR of ProFET features), which are all explained more below. Naive method reflects the prior probability of GO terms and BLAST-KNN makes use of homology based inference for function prediction. Amino acid trigram has been used as one component by Jones-UCL group, which achieved the first place in CAFA1 (Cozzetto et al., 2013). ProFET has been used in various function prediction task, LR-Interpro makes use of rich domain, family and motif information. These components from different information should be informative and complement to each other.
3.3.1 Naive: GO term frequency
3.3.2 BLAST-KNN (B-K): sequence alignment
3.3.3 LR-3mer: amino acid trigram
3.3.4 LR-InterPro: protein families, domains and motifs
InterPro (Mitchell et al., 2015) combines 14 different protein and domain family databases, including Pfam (Sonnhammer et al., 1997), CATH-Gene3D (Sillitoe et al., 2015), CCD (Marchler-Bauer et al., 2015) and SUPERFAMILY (de Lima et al., 2010), so covering a large number of protein families, domains and motifs in sequences. We run InterProScan (http://www.ebi.ac.uk/interpro/interproscan.html) over a sequence in D, resulting in a binary vector with 33 879 features, and then use this vector as an instance of training logistic regression classifier of each GO term.
3.3.5 LR-ProFET: sequence features including biophysical properties
ProFET (https://github.com/ddofer/ProFET) (Ofer and Linial, 2015) is a software toolkit extracting features from protein sequence for function prediction. These features can be divided into six categories: (i) Biophysical quantitative properties; (ii) Letter-based features; (iii) Local potential features; (iv) Information based statistics; (v) AA scale-based features; and (vi) Transformed CTD features. We run ProFET over a sequence in D to extract these features, resulting in a vector of 1170 features which is used as an input to train logistic regression classifier of each GO term.
3.4 GOLabeler (with three steps)
3.4.1 Step 1: Generate candidate GO terms
For a query protein, we run five component methods to have predicted GO terms, and after choosing the top-k predicted GO terms from each component, merge them together as the candidate GO terms (we used k = 30 in our experiments; See Section 4.3). Note that reducing k is to focus on the most relevant GO terms to query protein and also reduce the computational burden of the model.
3.4.2 Step 2: Generate features for ranking GO terms
We then generate features of the query protein by using the scores (of each of the candidate GO terms) predicted by all five component methods, resulting in a 5-dimensional feature vector for each pair of a GO term and one query protein. Note that all score values are between 0 and 1.
3.4.3 Step 3: Rank GO terms by learning to rank (LTR)
Finally, we use LTR to rank all candidate GO terms of each query protein. Note that all proteins in the training data and their candidate GO terms are used for training the LTR model. LTR can effectively integrate multiple sequence-based evidence for AFP of no-knowledge proteins in the framework of multilabel classification.
3.5 Competing methods
In our experiments, we compare GOLabeler with five methods: three ensemble approaches with the same component outputs as GOLabeler: one vote, weighted voting (WV) and consensus [which have been often used in other AFP work, e.g. (Vidulin et al., 2016)], and two existing methods, BLAST (Altschul et al., 1997) and GoFDR (Gong et al., 2016). We note that GoFDR was a top performer of CAFA2.
3.5.1 One vote
3.5.2 Weighted voting (WV)
3.5.3 Consensus
3.5.4 BLAST
3.5.5 GoFDR
Among the top performance methods in CAFA2, GoFDR is only the method having available source code (http://gofdr.tianlab.cn), and so we choose GoFDR as a competing method (Gong et al., 2016). For a query protein, GoFDR run BLAST or PSI-BLAST to obtain multiple sequence alignment (MSA) over the query sequence and find functionally discriminating residues (FDR) of each GO term in the MSA which are used to generate a position-specific scoring matrix (PSSM). GoFDR uses the PSSM to compute the score between the query protein and a GO term.
4 Experiments
4.1 Data
Data collection approximately followed the corresponding part of CAFA1 (Radivojac et al., 2013) and CAFA2 (Jiang et al., 2016):
Protein sequences
We downloaded the FASTA-format files of all proteins from UniProt (http://www.uniprot.org/downloads) (The UniProt Consortium, 2015).
GO terms
We downloaded protein function annotation from SwissProt (http://www.uniprot.org/downloads) (Boutet et al., 2016), GOA (http://www.ebi.ac.uk/GOA) (Huntley et al., 2015), and GO (http://geneontology.org/page/download-annotations) (Ashburner et al., 2000) in October 2016. Out of them we extracted all experimental annotations in: ‘EXP’, ‘IDA’, ‘IPI’, ‘IMP’, ‘IGI’, ‘IEP’, ‘TAS’ or ‘IC’, and then merged them to form a full annotation dataset (Note that SwissProt did not have annotation dates and so we downloaded data of SwissProt in January 2016 and January 2015 also).
We then generated the following four datasets, which are mainly separated by the time stamps that proteins are annotated.
Training: training for components
All data annotated in 2014 or before.
LTR1: training for LTR
Among the data experimentally annotated in 2015 and not before 2015, no-knowledge proteins.
LTR2: training for LTR
Among the data experimentally annotated in 2015 and not before 2015, limited-knowledge proteins.
Testing: testing for competing methods
All data experimentally annotated in 2016 (January to October of 2016, since we downloaded the data in October 2016) and not before 2016.
Note that this time-series way of separating training and testing data is the same as CAFA. Also we used the same target species as CAFA3, an ongoing challenge for AFP, in LTR1, LTR2 and Testing. Table 1 shows the number of proteins in the four datasets. We used Testing (or we call benchmark) as the testset to examine the performance of competing methods.
Data statistics (# proteins) on species with at least ten proteins for one domain of GO for each of the four datasets
| Species | Training | LTR1 | LTR2 | Testing | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| HUMAN (Homo sapiens) | 9087 | 11019 | 15 977 | 150 | 216 | 718 | 261 | 454 | 161 | 42 | 64 | 67 |
| MOUSE (Mus musculus) | 5697 | 9262 | 8770 | 169 | 339 | 237 | 140 | 137 | 175 | 100 | 235 | 146 |
| DROME (Drosophila melanogaster) | 4646 | 10 778 | 7609 | 43 | 579 | 172 | 132 | 224 | 327 | 125 | 339 | 188 |
| ARATH (Arabidopsis thaliana) | 3857 | 7238 | 8492 | 84 | 198 | 140 | 195 | 114 | 70 | 84 | 180 | 128 |
| DANRE (Danio rerio) | 2173 | 8374 | 1500 | 76 | 722 | 159 | 108 | 56 | 50 | 66 | 380 | 27 |
| RAT (Rattus norvegicus) | 4199 | 5128 | 4 459 | 121 | 185 | 178 | 56 | 70 | 106 | 31 | 88 | 88 |
| DICDI (Dictyostelium discoideum) | 414 | 921 | 803 | 17 | 52 | 25 | 16 | 15 | 19 | 16 | 26 | 12 |
| All species (not only the above) | 45 543 | 77 170 | 71 388 | 724 | 2387 | 1679 | 1081 | 1206 | 956 | 497 | 1340 | 770 |
| Species | Training | LTR1 | LTR2 | Testing | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| HUMAN (Homo sapiens) | 9087 | 11019 | 15 977 | 150 | 216 | 718 | 261 | 454 | 161 | 42 | 64 | 67 |
| MOUSE (Mus musculus) | 5697 | 9262 | 8770 | 169 | 339 | 237 | 140 | 137 | 175 | 100 | 235 | 146 |
| DROME (Drosophila melanogaster) | 4646 | 10 778 | 7609 | 43 | 579 | 172 | 132 | 224 | 327 | 125 | 339 | 188 |
| ARATH (Arabidopsis thaliana) | 3857 | 7238 | 8492 | 84 | 198 | 140 | 195 | 114 | 70 | 84 | 180 | 128 |
| DANRE (Danio rerio) | 2173 | 8374 | 1500 | 76 | 722 | 159 | 108 | 56 | 50 | 66 | 380 | 27 |
| RAT (Rattus norvegicus) | 4199 | 5128 | 4 459 | 121 | 185 | 178 | 56 | 70 | 106 | 31 | 88 | 88 |
| DICDI (Dictyostelium discoideum) | 414 | 921 | 803 | 17 | 52 | 25 | 16 | 15 | 19 | 16 | 26 | 12 |
| All species (not only the above) | 45 543 | 77 170 | 71 388 | 724 | 2387 | 1679 | 1081 | 1206 | 956 | 497 | 1340 | 770 |
Data statistics (# proteins) on species with at least ten proteins for one domain of GO for each of the four datasets
| Species | Training | LTR1 | LTR2 | Testing | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| HUMAN (Homo sapiens) | 9087 | 11019 | 15 977 | 150 | 216 | 718 | 261 | 454 | 161 | 42 | 64 | 67 |
| MOUSE (Mus musculus) | 5697 | 9262 | 8770 | 169 | 339 | 237 | 140 | 137 | 175 | 100 | 235 | 146 |
| DROME (Drosophila melanogaster) | 4646 | 10 778 | 7609 | 43 | 579 | 172 | 132 | 224 | 327 | 125 | 339 | 188 |
| ARATH (Arabidopsis thaliana) | 3857 | 7238 | 8492 | 84 | 198 | 140 | 195 | 114 | 70 | 84 | 180 | 128 |
| DANRE (Danio rerio) | 2173 | 8374 | 1500 | 76 | 722 | 159 | 108 | 56 | 50 | 66 | 380 | 27 |
| RAT (Rattus norvegicus) | 4199 | 5128 | 4 459 | 121 | 185 | 178 | 56 | 70 | 106 | 31 | 88 | 88 |
| DICDI (Dictyostelium discoideum) | 414 | 921 | 803 | 17 | 52 | 25 | 16 | 15 | 19 | 16 | 26 | 12 |
| All species (not only the above) | 45 543 | 77 170 | 71 388 | 724 | 2387 | 1679 | 1081 | 1206 | 956 | 497 | 1340 | 770 |
| Species | Training | LTR1 | LTR2 | Testing | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| HUMAN (Homo sapiens) | 9087 | 11019 | 15 977 | 150 | 216 | 718 | 261 | 454 | 161 | 42 | 64 | 67 |
| MOUSE (Mus musculus) | 5697 | 9262 | 8770 | 169 | 339 | 237 | 140 | 137 | 175 | 100 | 235 | 146 |
| DROME (Drosophila melanogaster) | 4646 | 10 778 | 7609 | 43 | 579 | 172 | 132 | 224 | 327 | 125 | 339 | 188 |
| ARATH (Arabidopsis thaliana) | 3857 | 7238 | 8492 | 84 | 198 | 140 | 195 | 114 | 70 | 84 | 180 | 128 |
| DANRE (Danio rerio) | 2173 | 8374 | 1500 | 76 | 722 | 159 | 108 | 56 | 50 | 66 | 380 | 27 |
| RAT (Rattus norvegicus) | 4199 | 5128 | 4 459 | 121 | 185 | 178 | 56 | 70 | 106 | 31 | 88 | 88 |
| DICDI (Dictyostelium discoideum) | 414 | 921 | 803 | 17 | 52 | 25 | 16 | 15 | 19 | 16 | 26 | 12 |
| All species (not only the above) | 45 543 | 77 170 | 71 388 | 724 | 2387 | 1679 | 1081 | 1206 | 956 | 497 | 1340 | 770 |
4.2 Performance evaluation measures
Practically we evaluated the top 100 GO terms predicted by competing methods for each GO domain (we used 100, since the number of GO terms per protein is clearly smaller than 100, and also simply the top GO terms are important).
4.3 Implementation and parameter settings
We processed the FASTA-format data by biopython (http://biopython.org/) and used sklearn (http://scikit-learn.org/stable/index.html) for running logistic regression and xgboost (Chen and Guestrin, 2016) to run LTR.
4.3.1 GOLabeler
BLAST-KNN
Ver. 2.3.0+ was used with default parameters for BLAST, except that blastdb was from all proteins in D and the number of iterations was one.
LTR
We used ‘rank: pairwise’ as the objective loss function in xgboost. Also the maximum depth of trees in MART (Multiple Additive Regression Trees) was set at 4, to avoid overfitting to the training data. We selected top 30 predictions from each component to be merged, since this number provided the best performance in five-fold cross validation over LTR training data (out of four values {10, 30, 50 and 70} tested).
4.3.2 Competing methods
BLAST
We used the same setting as BLAST-KNN.
GoFDR
We ran GoFDR over all required data [i.e. all annotation without ‘IEA’ or ‘RCA’ evidence and some annotations with IEA evidence before 2016 from GOA (Huntley et al., 2015)].
4.4 Results
We resampled the test dataset with replacement 100 times (bootstrap with replacement) to make the experiment reliable. We used not only the three performance evaluation measure, but also paired t-test to statistically evaluate the performance difference between the best performance method (in boldface in tables) and all other methods. The result was considered significant if P value was smaller than 0.05. Then in tables, the best performance value is underlined if the value is statistically significant (see the supplementary materials for detailed P values).
4.4.1 Comparison with component methods
We first compare the five component methods, the results being shown in Table 2, where out of the five methods, the best values in each column are in italics. Out of the nine (=three evaluation criteria times three domains) comparison settings, LR-InterPro achieved the best five times, being followed by BLAST-KNN which achieved the best three times. This suggests that LR-InterPro and BLAST-KNN are the two best component methods, which means that domain, family, motif and homology information are very informative. The other three methods are less accurate, while their high performance could be found in some specific cases: LR-3mer achieved the highest Fmax in CCO. We then examined the performance of GOLabeler trained by LTR1, the results being shown in the same table, where GOLabeler with only two components was also checked as well as GOLabeler (with all five components), which are called GOL (B + I) and GOL (All), respectively. The table shows that GOL (All) outperformed all competing methods in eight out of nine evaluation settings. For example, GOL (All) achieved the highest Fmax of 0.580 in MFO, followed by GOL (B + I) of 0.578, BLAST-KNN of 0.573 and LR-InterPro of 0.556. This result indicates the advantage of incorporating all component methods in GOLabeler, compared with using only a smaller number of components. Another finding was that among MFO, BPO and CCO, BPO is the hardest task in AFP (this is consistent with the results of CAFA). For example, the best AUPR of GOL (All) was 0.546 and 0.700 for MFO and CCO, respectively, while it was only 0.225 for BPO, implying that sequences are the limited information for BPO in AFP. Hereafter, we show the result by GOL (All) as that by GOLabeler.
Performance comparison with component methods
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| Naive | 0.141 | 0.151 | 0.591 | 0.242 | 0.299 | 0.653 | 7.684 | 15.965 | 6.535 |
| B-K | 0.452 | 0.192 | 0.558 | 0.573 | 0.339 | 0.620 | 5.157 | 15.713 | 5.647 |
| LR-3mer | 0.144 | 0.152 | 0.600 | 0.255 | 0.301 | 0.664 | 7.587 | 15.934 | 6.415 |
| LR-InterPro | 0.536 | 0.198 | 0.636 | 0.556 | 0.351 | 0.654 | 5.248 | 15.655 | 5.783 |
| LR-ProFET | 0.173 | 0.096 | 0.550 | 0.330 | 0.265 | 0.633 | 7.831 | 17.030 | 6.380 |
| GOL (B+I) | 0.538 | 0.173 | 0.657 | 0.578 | 0.352 | 0.665 | 5.126 | 15.225 | 5.439 |
| GOL (All) | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| Naive | 0.141 | 0.151 | 0.591 | 0.242 | 0.299 | 0.653 | 7.684 | 15.965 | 6.535 |
| B-K | 0.452 | 0.192 | 0.558 | 0.573 | 0.339 | 0.620 | 5.157 | 15.713 | 5.647 |
| LR-3mer | 0.144 | 0.152 | 0.600 | 0.255 | 0.301 | 0.664 | 7.587 | 15.934 | 6.415 |
| LR-InterPro | 0.536 | 0.198 | 0.636 | 0.556 | 0.351 | 0.654 | 5.248 | 15.655 | 5.783 |
| LR-ProFET | 0.173 | 0.096 | 0.550 | 0.330 | 0.265 | 0.633 | 7.831 | 17.030 | 6.380 |
| GOL (B+I) | 0.538 | 0.173 | 0.657 | 0.578 | 0.352 | 0.665 | 5.126 | 15.225 | 5.439 |
| GOL (All) | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
Note: B-K, BLAST-KNN; GOL (B + I), only BLAST-KNN and LR-InterPro were used for components in GOLabeler; GOL (All), all five components are used in GOLabeler.
Performance comparison with component methods
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| Naive | 0.141 | 0.151 | 0.591 | 0.242 | 0.299 | 0.653 | 7.684 | 15.965 | 6.535 |
| B-K | 0.452 | 0.192 | 0.558 | 0.573 | 0.339 | 0.620 | 5.157 | 15.713 | 5.647 |
| LR-3mer | 0.144 | 0.152 | 0.600 | 0.255 | 0.301 | 0.664 | 7.587 | 15.934 | 6.415 |
| LR-InterPro | 0.536 | 0.198 | 0.636 | 0.556 | 0.351 | 0.654 | 5.248 | 15.655 | 5.783 |
| LR-ProFET | 0.173 | 0.096 | 0.550 | 0.330 | 0.265 | 0.633 | 7.831 | 17.030 | 6.380 |
| GOL (B+I) | 0.538 | 0.173 | 0.657 | 0.578 | 0.352 | 0.665 | 5.126 | 15.225 | 5.439 |
| GOL (All) | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| Naive | 0.141 | 0.151 | 0.591 | 0.242 | 0.299 | 0.653 | 7.684 | 15.965 | 6.535 |
| B-K | 0.452 | 0.192 | 0.558 | 0.573 | 0.339 | 0.620 | 5.157 | 15.713 | 5.647 |
| LR-3mer | 0.144 | 0.152 | 0.600 | 0.255 | 0.301 | 0.664 | 7.587 | 15.934 | 6.415 |
| LR-InterPro | 0.536 | 0.198 | 0.636 | 0.556 | 0.351 | 0.654 | 5.248 | 15.655 | 5.783 |
| LR-ProFET | 0.173 | 0.096 | 0.550 | 0.330 | 0.265 | 0.633 | 7.831 | 17.030 | 6.380 |
| GOL (B+I) | 0.538 | 0.173 | 0.657 | 0.578 | 0.352 | 0.665 | 5.126 | 15.225 | 5.439 |
| GOL (All) | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
Note: B-K, BLAST-KNN; GOL (B + I), only BLAST-KNN and LR-InterPro were used for components in GOLabeler; GOL (All), all five components are used in GOLabeler.
4.4.2 Comparison with other ensemble techniques and also performance improvement by making training size larger
We examined the performance of recent ensemble techniques with the same five component methods and also the performance improvement by increasing the training data for ensemble learning, i.e. weighted voting and GOLabeler. Table 3 shows the result summary under nine experimental settings. When we used LTR1 only (GOL-L1), GOLabeler achieved the best performance among the competing methods. Weighted voting was the next best, while the performance of one vote was lowest in all nine settings, implying that choosing one component would not work well. By adding LTR2, the performance of GOLabeler (GOL-L1 + 2) was more pronounced, achieving the best in all nine settings, except only one, while the performance of weighted voting was increased in only five out of all nine cases. This means that GOLabeler can take the advantage of using a larger size of data more effectively than weighted voting. Also this implies that the performance of GOLabeler can be rather easily improved further by increasing the annotation data in the future. Hereafter we show the results of GOLabeler and weighted voting with all training data for LTR (both LTR1 and LTR2) as those by GOLabeler and weighted voting, respectively.
Performance comparison with ensemble techniques and also improvement by adding LTR2 to LTR1
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| One vote | 0.423 | 0.193 | 0.651 | 0.543 | 0.335 | 0.682 | 6.116 | 16.237 | 5.883 |
| WV-L1 | 0.530 | 0.230 | 0.694 | 0.571 | 0.368 | 0.692 | 5.119 | 15.117 | 5.516 |
| WV-L1 + 2 | 0.530 | 0.232 | 0.694 | 0.571 | 0.370 | 0.692 | 5.114 | 15.101 | 5.506 |
| Consensus | 0.454 | 0.222 | 0.670 | 0.543 | 0.360 | 0.692 | 5.687 | 15.510 | 5.660 |
| GOL-L1 | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
| GOL-L1 + 2 | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| One vote | 0.423 | 0.193 | 0.651 | 0.543 | 0.335 | 0.682 | 6.116 | 16.237 | 5.883 |
| WV-L1 | 0.530 | 0.230 | 0.694 | 0.571 | 0.368 | 0.692 | 5.119 | 15.117 | 5.516 |
| WV-L1 + 2 | 0.530 | 0.232 | 0.694 | 0.571 | 0.370 | 0.692 | 5.114 | 15.101 | 5.506 |
| Consensus | 0.454 | 0.222 | 0.670 | 0.543 | 0.360 | 0.692 | 5.687 | 15.510 | 5.660 |
| GOL-L1 | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
| GOL-L1 + 2 | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
Note: WV-L1, Weighted voting with LTR1 only; WV-L1 + 2, Weighted voting with LTR1 and LTR2; GOL-L1, GOlabeler with LTR1 only; GOL-L1 + 2, GOLabeler with LTR1 and LTR2.
Performance comparison with ensemble techniques and also improvement by adding LTR2 to LTR1
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| One vote | 0.423 | 0.193 | 0.651 | 0.543 | 0.335 | 0.682 | 6.116 | 16.237 | 5.883 |
| WV-L1 | 0.530 | 0.230 | 0.694 | 0.571 | 0.368 | 0.692 | 5.119 | 15.117 | 5.516 |
| WV-L1 + 2 | 0.530 | 0.232 | 0.694 | 0.571 | 0.370 | 0.692 | 5.114 | 15.101 | 5.506 |
| Consensus | 0.454 | 0.222 | 0.670 | 0.543 | 0.360 | 0.692 | 5.687 | 15.510 | 5.660 |
| GOL-L1 | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
| GOL-L1 + 2 | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| One vote | 0.423 | 0.193 | 0.651 | 0.543 | 0.335 | 0.682 | 6.116 | 16.237 | 5.883 |
| WV-L1 | 0.530 | 0.230 | 0.694 | 0.571 | 0.368 | 0.692 | 5.119 | 15.117 | 5.516 |
| WV-L1 + 2 | 0.530 | 0.232 | 0.694 | 0.571 | 0.370 | 0.692 | 5.114 | 15.101 | 5.506 |
| Consensus | 0.454 | 0.222 | 0.670 | 0.543 | 0.360 | 0.692 | 5.687 | 15.510 | 5.660 |
| GOL-L1 | 0.546 | 0.225 | 0.700 | 0.580 | 0.370 | 0.687 | 5.077 | 15.177 | 5.518 |
| GOL-L1 + 2 | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
Note: WV-L1, Weighted voting with LTR1 only; WV-L1 + 2, Weighted voting with LTR1 and LTR2; GOL-L1, GOlabeler with LTR1 only; GOL-L1 + 2, GOLabeler with LTR1 and LTR2.
4.4.3 Comparison with BLAST and GoFDR
Table 4 shows the performance of BLAST and GoFDR. GOLabeler outperformed both BLAST and GoFDR in all experimental settings, except only one, being statistically significant. GoFDR is a method that achieved the top performance in CAFA2, demonstrating the high performance of GOLabeler, even compared with high performers in CAFA.
Performance comparison with BLAST and GoFDR
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| BLAST | 0.263 | 0.071 | 0.311 | 0.435 | 0.262 | 0.513 | 7.223 | 17.358 | 6.848 |
| GoFDR | 0.424 | 0.183 | 0.503 | 0.535 | 0.322 | 0.587 | 6.075 | 16.909 | 5.424 |
| GOLabeler | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| BLAST | 0.263 | 0.071 | 0.311 | 0.435 | 0.262 | 0.513 | 7.223 | 17.358 | 6.848 |
| GoFDR | 0.424 | 0.183 | 0.503 | 0.535 | 0.322 | 0.587 | 6.075 | 16.909 | 5.424 |
| GOLabeler | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
Performance comparison with BLAST and GoFDR
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| BLAST | 0.263 | 0.071 | 0.311 | 0.435 | 0.262 | 0.513 | 7.223 | 17.358 | 6.848 |
| GoFDR | 0.424 | 0.183 | 0.503 | 0.535 | 0.322 | 0.587 | 6.075 | 16.909 | 5.424 |
| GOLabeler | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| BLAST | 0.263 | 0.071 | 0.311 | 0.435 | 0.262 | 0.513 | 7.223 | 17.358 | 6.848 |
| GoFDR | 0.424 | 0.183 | 0.503 | 0.535 | 0.322 | 0.587 | 6.075 | 16.909 | 5.424 |
| GOLabeler | 0.549 | 0.236 | 0.697 | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 |
4.4.4 Performance over ‘difficult’ proteins
The no-knowledge test dataset can be divided into two datasets: easy and difficult proteins, due to the sequence identity of 60% to the most similar known protein in the training data (Jiang et al., 2016). The size of difficult proteins in the test dataset is 370 in MF, 870 in BP and 532 in CC, respectively. As mentioned in Introduction, AFP for difficult proteins is especially important, since homology-based methods, such as BLAST-KNN, are unable to predict the functions of difficult proteins. Table 5 shows the summary of results over difficult proteins in the test dataset, compared with two best component methods, BLAST-KNN and LR-InterPro, and two ensemble methods, weighted voting and consensus. From the table, we can find that GOLabeler could outperform all competing methods in all nine settings, meaning that the performance advantage of this case was much clearer than the case of using both difficult and easy proteins. We note that this result indicates that GOLabeler is particularly useful for AFP of difficult proteins, for which effective methods currently do not exist.
Performance comparison over ‘difficult’ proteins
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| B-K | 0.400 | 0.203 | 0.510 | 0.553 | 0.350 | 0.603 | 5.194 | 15.410 | 5.658 |
| LR-InterPro | 0.505 | 0.217 | 0.639 | 0.534 | 0.363 | 0.663 | 5.306 | 15.074 | 5.581 |
| Consensus | 0.407 | 0.239 | 0.659 | 0.517 | 0.368 | 0.699 | 5.987 | 15.217 | 5.678 |
| WV | 0.497 | 0.248 | 0.702 | 0.547 | 0.379 | 0.701 | 5.264 | 14.612 | 5.423 |
| GOLabeler | 0.516 | 0.258 | 0.704 | 0.567 | 0.382 | 0.706 | 5.087 | 14.538 | 5.344 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| B-K | 0.400 | 0.203 | 0.510 | 0.553 | 0.350 | 0.603 | 5.194 | 15.410 | 5.658 |
| LR-InterPro | 0.505 | 0.217 | 0.639 | 0.534 | 0.363 | 0.663 | 5.306 | 15.074 | 5.581 |
| Consensus | 0.407 | 0.239 | 0.659 | 0.517 | 0.368 | 0.699 | 5.987 | 15.217 | 5.678 |
| WV | 0.497 | 0.248 | 0.702 | 0.547 | 0.379 | 0.701 | 5.264 | 14.612 | 5.423 |
| GOLabeler | 0.516 | 0.258 | 0.704 | 0.567 | 0.382 | 0.706 | 5.087 | 14.538 | 5.344 |
Note: B-K, BLAST-KNN; WV, Weighted voting.
Performance comparison over ‘difficult’ proteins
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| B-K | 0.400 | 0.203 | 0.510 | 0.553 | 0.350 | 0.603 | 5.194 | 15.410 | 5.658 |
| LR-InterPro | 0.505 | 0.217 | 0.639 | 0.534 | 0.363 | 0.663 | 5.306 | 15.074 | 5.581 |
| Consensus | 0.407 | 0.239 | 0.659 | 0.517 | 0.368 | 0.699 | 5.987 | 15.217 | 5.678 |
| WV | 0.497 | 0.248 | 0.702 | 0.547 | 0.379 | 0.701 | 5.264 | 14.612 | 5.423 |
| GOLabeler | 0.516 | 0.258 | 0.704 | 0.567 | 0.382 | 0.706 | 5.087 | 14.538 | 5.344 |
| AUPR | Fmax | Smin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| B-K | 0.400 | 0.203 | 0.510 | 0.553 | 0.350 | 0.603 | 5.194 | 15.410 | 5.658 |
| LR-InterPro | 0.505 | 0.217 | 0.639 | 0.534 | 0.363 | 0.663 | 5.306 | 15.074 | 5.581 |
| Consensus | 0.407 | 0.239 | 0.659 | 0.517 | 0.368 | 0.699 | 5.987 | 15.217 | 5.678 |
| WV | 0.497 | 0.248 | 0.702 | 0.547 | 0.379 | 0.701 | 5.264 | 14.612 | 5.423 |
| GOLabeler | 0.516 | 0.258 | 0.704 | 0.567 | 0.382 | 0.706 | 5.087 | 14.538 | 5.344 |
Note: B-K, BLAST-KNN; WV, Weighted voting.
4.4.5 Case study
Finally, we show a specific example of the results obtained by GOLabeler and other competing methods, to illustrate the real effect of the performance difference on annotating GO to unknown proteins. Table 6 shows the list of predicted GO terms (The GO terms by each method are determined by its own cut-off value to achieve the best value of Fmax) of MFO for a protein, Wor4p (Uniprot Symbol: Q5ADX8), which is, in ground-truth, associated with 12 GO terms in MFO. Also Figure 2 shows the directed acyclic graph by the 12 GO terms associated with Q5ADX8. Note that Q5ADX8 is a difficult protein. There are no homologous proteins of Q5ADX8 (set the cut-off of e-value at 0.001), by which BLAST-KNN was unable to predict any GO terms. Naive predicted three GO terms, out of which only one very generally term, GO: 0005488 (Binding), was correct. By checking the InterPro database, we found that Q5ADX8 matches Zinc finger C2H2-type domain (IPR013087) (https://www.ebi.ac.uk/interpro/protein/Q5ADX8), which was associated with GO: 0003676 (nucleic acid binding) (http://www.ebi.ac.uk/interpro/entry/IPR013087). This was exactly what LR-InterPro predicted. Compared with Naive method, LR-InterPro, LR-3mer and LR-ProFET could make more correct annotations particularly for more specific terms, such as GO: 0003676 (nucleic acid binding). The prediction by weighted voting was also at the same level of specificity as the three component methods (The poor performance of weighted voting might be caused by BLAST-KNN, which was unable to predict any GO term). In fact, the predicted GO terms by LR-InterPro and weighted voting were all correct, but the number of the predicted GO terms was only four and five, respectively. On the other hand, GOLabeler predicted nine terms, out of which eight were correct. Note that the only wrong predicted GO term GO: 0005515 (protein binding) was a very general term, which was also predicted by Naive, LR-3mer and LR-PROFET. More importantly, as shown in Figure 2, the prediction by GOLabeler was most specific. For example, even GO: 0044212 (transcription regulatory region DNA binding), which is next to the end node in Figure 2, was correctly annotated. From this result, we can see that the performance advantage of GOLabeler results in sizeable differences in quality of real function annotation.
Predicted GO terms of Q5ADX8 in MFO by GOLabeler and competing methods
| Method | Predicted GO terms |
|---|---|
| Naive | GO: 0005488, GO: 0003824, GO: 0005515 |
| GoFDR | GO: 0005488, GO: 0043169, GO: 0043167, GO:00046872, GO: 0097159, GO: 0003676, GO: 1901363, GO: 0003674 |
| B-K | |
| LR-3mer | GO: 0005488, GO: 0005515, GO: 0003824, GO: 0097159, GO: 1901363, GO: 0003676 |
| LR-InterPro | GO: 0005488, GO: 0003677, GO: 1901363, GO: 0097159, GO: 0003676 |
| LR-ProFET | GO: 0003676, GO: 0097159, GO: 1901363, GO: 0005488, GO: 0003723, GO: 0003677, GO: 0001010, GO: 0005515 |
| WV | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676 |
| GOLabeler | GO: 0005488, GO: 0003676, GO: 0097159, GO: 1901363, GO: 0003677, GO: 0005515, GO: 0044212, GO: 0000975, GO: 0001067 |
| Ground truth | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676, GO: 0001067, GO: 0003677, GO: 0000975, GO: 0003690, GO: 0043565 |
| GO: 0044212, GO: 1990837, GO: 0000976 |
| Method | Predicted GO terms |
|---|---|
| Naive | GO: 0005488, GO: 0003824, GO: 0005515 |
| GoFDR | GO: 0005488, GO: 0043169, GO: 0043167, GO:00046872, GO: 0097159, GO: 0003676, GO: 1901363, GO: 0003674 |
| B-K | |
| LR-3mer | GO: 0005488, GO: 0005515, GO: 0003824, GO: 0097159, GO: 1901363, GO: 0003676 |
| LR-InterPro | GO: 0005488, GO: 0003677, GO: 1901363, GO: 0097159, GO: 0003676 |
| LR-ProFET | GO: 0003676, GO: 0097159, GO: 1901363, GO: 0005488, GO: 0003723, GO: 0003677, GO: 0001010, GO: 0005515 |
| WV | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676 |
| GOLabeler | GO: 0005488, GO: 0003676, GO: 0097159, GO: 1901363, GO: 0003677, GO: 0005515, GO: 0044212, GO: 0000975, GO: 0001067 |
| Ground truth | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676, GO: 0001067, GO: 0003677, GO: 0000975, GO: 0003690, GO: 0043565 |
| GO: 0044212, GO: 1990837, GO: 0000976 |
Note: Correctly predicted GO Terms are in bold face.
WV, Weighted voting.
Predicted GO terms of Q5ADX8 in MFO by GOLabeler and competing methods
| Method | Predicted GO terms |
|---|---|
| Naive | GO: 0005488, GO: 0003824, GO: 0005515 |
| GoFDR | GO: 0005488, GO: 0043169, GO: 0043167, GO:00046872, GO: 0097159, GO: 0003676, GO: 1901363, GO: 0003674 |
| B-K | |
| LR-3mer | GO: 0005488, GO: 0005515, GO: 0003824, GO: 0097159, GO: 1901363, GO: 0003676 |
| LR-InterPro | GO: 0005488, GO: 0003677, GO: 1901363, GO: 0097159, GO: 0003676 |
| LR-ProFET | GO: 0003676, GO: 0097159, GO: 1901363, GO: 0005488, GO: 0003723, GO: 0003677, GO: 0001010, GO: 0005515 |
| WV | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676 |
| GOLabeler | GO: 0005488, GO: 0003676, GO: 0097159, GO: 1901363, GO: 0003677, GO: 0005515, GO: 0044212, GO: 0000975, GO: 0001067 |
| Ground truth | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676, GO: 0001067, GO: 0003677, GO: 0000975, GO: 0003690, GO: 0043565 |
| GO: 0044212, GO: 1990837, GO: 0000976 |
| Method | Predicted GO terms |
|---|---|
| Naive | GO: 0005488, GO: 0003824, GO: 0005515 |
| GoFDR | GO: 0005488, GO: 0043169, GO: 0043167, GO:00046872, GO: 0097159, GO: 0003676, GO: 1901363, GO: 0003674 |
| B-K | |
| LR-3mer | GO: 0005488, GO: 0005515, GO: 0003824, GO: 0097159, GO: 1901363, GO: 0003676 |
| LR-InterPro | GO: 0005488, GO: 0003677, GO: 1901363, GO: 0097159, GO: 0003676 |
| LR-ProFET | GO: 0003676, GO: 0097159, GO: 1901363, GO: 0005488, GO: 0003723, GO: 0003677, GO: 0001010, GO: 0005515 |
| WV | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676 |
| GOLabeler | GO: 0005488, GO: 0003676, GO: 0097159, GO: 1901363, GO: 0003677, GO: 0005515, GO: 0044212, GO: 0000975, GO: 0001067 |
| Ground truth | GO: 0005488, GO: 0097159, GO: 1901363, GO: 0003676, GO: 0001067, GO: 0003677, GO: 0000975, GO: 0003690, GO: 0043565 |
| GO: 0044212, GO: 1990837, GO: 0000976 |
Note: Correctly predicted GO Terms are in bold face.
WV, Weighted voting.
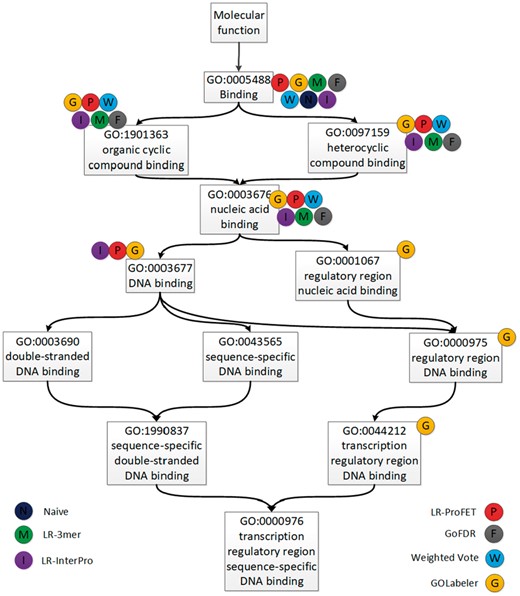
Predicted GO terms of Q5ADX8 in DAG of MFO by different methods
4.5 Computational efficiency
We implemented GOLabeler on a server with Intel(R) Core(TM) i7-6700 K 4.00 GHz CPU and 64GB RAM. Training all component methods and ranking models took around 10 days. During prediction, most computation was spent on BLAST-KNN. Given a thousand new proteins, annotating GO took only less then 6 h.
5 Conclusion and discussion
Sequence-based large-scale AFP (SAFP) for proteins, particularly for difficult proteins, is an important problem, especially with three challenging issues: (i) structured ontology, (ii) many labels per protein and (iii) large variation in the number of GO terms per protein. For this problem, we have proposed GOLabeler, addressing the three issues: (i) by using all corresponding GO terms in the DAG structure of GO, (ii) by learning to rank (LTR), a very effective multilabel classification framework and also (iii) by LTR, which allows not to select the number of GO terms per protein. Our thorough and extensive experiments show the clear advantage of GOLabeler in predictive performance from many viewpoints over state-of-the-art techniques in SAFP and ensemble approaches. Diverse information from sequences is very useful for SAFP. GOLabeler currently integrates five classifiers as components which are from GO term frequency, sequence homology, trigram, motifs and biophysical properties, and so on. The framework of GOLabeler or LTR is flexible and allows to incorporate any classifiers we can use, implying that further performance improvement would be possible by adding more information from sequences. Also this could be done for not only SAFP but also more general AFP. This would be interesting future work. Also possible future work would be to develop GOLabeler to improve the performance of AFP for specific species, especially for those with very few proteins, for which current AFP methods do not necessarily work sufficiently. Finally, with the development of structure prediction methods, it will be increasingly important to integrate protein sequence with predicted structure for function prediction.
Funding
This work has been supported in part by the National Natural Science Foundation of China (NOs. 61572139 and 31601074), MEXT KAKENHI 16H02868, JST: ACCEL, Tekes (currently Business Finland): FiDiPro, Academy of Finland: AIPSE programme, and the Open Fund of Shanghai Key Laboratory of Intelligent Information Processing (No. IIPL-2016-005).
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}