





Abstract
Next-generation sequencing (NGS) technologies have led to the accumulation of high-throughput sequence data from various organisms in biology. To apply gene annotation of organellar genomes for various organisms, more optimized tools for functional gene annotation are required. Almost all gene annotation tools are mainly focused on the chloroplast genome of land plants or the mitochondrial genome of animals. We have developed a web application AGORA for the fast, user-friendly and improved annotations of organellar genomes. Annotator for Genes of Organelle from the Reference sequence Analysis (AGORA) annotates genes based on a basic local alignment search tool (BLAST)-based homology search and clustering with selected reference sequences from the NCBI database or user-defined uploaded data. AGORA can annotate the functional genes in almost all mitochondrion and plastid genomes of eukaryotes. The gene annotation of a genome with an exon–intron structure within a gene or inverted repeat region is also available. It provides information of start and end positions of each gene, BLAST results compared with the reference sequence and visualization of gene map by OGDRAW.
Users can freely use the software, and the accessible URL is https://bigdata.dongguk.edu/gene_project/AGORA/. The main module of the tool is implemented by the python and php, and the web page is built by the HTML and CSS to support all browsers.
Supplementary data are available at Bioinformatics online.
1 Introduction
Next-Generation Sequencing (NGS) technologies have been in a remarkable era for big data in biology and have led to the accumulation of high-throughput genome data in biology. Even though biologists can obtain high-throughput genome data from various organisms, genome assembly and functional gene annotation are still challenging problems. Especially, this study was focused on the development of a new application for gene annotation of the organellar genome in almost all eukaryotes. Five automated tools for gene annotation of organellar genome are available: DOGMA (Wyman et al., 2004), GeSeq (Tillich et al., 2017), CpGAVAS (Liu et al., 2012), Mitofy (Alverson et al., 2010) and Verdant (McKain et al., 2017). Almost all tools for automated gene annotation have been optimized mainly for the chloroplast genome of land plants. Only two tools have been developed for the mitochondrial genome: Mitofy for plants and DOGMA for animals. Moreover, almost all tools have restricted to select reference sequences. They are not allowed to use the customized data as a reference sequence or change the reference in other eukaryotes. The results of existing programs were matched with few genes or are insufficient to annotate genes for plastid and mitochondrial genomes in protist.
We introduce annotator for genes of organelle from the reference sequence analysis (AGORA), a web-based gene annotation tool for organellar genomes. The program was designed for gene annotation based on a basic local alignment search tool (BLAST) search compared with a user-selected reference sequence. AGORA was tested for almost all mitochondrion and plastid genomes of eukaryotes. The program is user-optimized for selected genomes and organism categories (i.e. plant, animal, algae or protist). The user change of the reference sequence allows the program to annotate all genes from the nuclear and nucleomorph genomes of eukaryotes or from the bacterial genome. The program produces high-quality results for annotated genes, even with a genome with an exon-intro structure within a gene or for an inverted repeat region. The program provides coordination information with start and end nucleotides for each gene that includes functional protein-coding gene, tRNAs and rRNAs, BLAST results of each gene information compared with the reference genome, visualization for gene content, and arrangement by ORDRAW (Lohse et al., 2013, 2007).
2 Implementation
The AGORA is a user-friendly web interface implemented by Javascript, php, Python and Bootstrap (Spurlock, 2013) framework. The core module is composed of three parts:
A Python-based script modules for gene annotating by maximizing sequence similarity from the references.
A web-based service modules to display the result and representing the image and formatted results.
A web-based service provider for managing the jobs for each users.
Among the three component steps, the core role is conducted in the first step, which is described in Figure 1. The overview describes the input and output formatted data and a running module for each step. The system requires several options, such as query sequence, reference sequences, genetic code and count of maximum matched genes. The description of user inputs is explained in the Supplementary Figure S1. The main goal of AGORA is to annotate genes from references. For reference sequences, users can insert the accession number from NCBI or user-defined reference sequences. In case of an accession number, the system automatically creates amino acid and nucleotide reference sequences. However, if the user wants to use user-defined reference sequences directly in the system, both amino acid and nucleotide sequences are required. The query is an assembled nucleotide sequence in the FASTA format. The genetic code is used to execute tblastn to search the translated nucleotide database with the protein query. The next step is an execution of the BLAST. Based on the references, two types of BLAST are executed. tblastn is run for amino acids of protein coding sequence with the genetic code, which is a user-defined option value (Supplementary Fig. S1) and blastn is executed for nucleotides of tRNAs and rRNAs. These two runs are shown in different line style in Figure 1, in which the solid line indicates the amino acids and the dotted line indicates the nucleotides. From these BLAST results, candidate genes can be identified by filtering the maximum number of matched genes. If the maximum value is set to 1, the result shows only the highstmatched candidate gene. However, if users wish to identify additional matched genes and analyzing exons and introns, the maximum value is set to higher than 1. During this filtering step, a GenBank formatted file is also created. AGORA provides not only the output of GenBank file format but also additional outputs, such as matched position of genes, BLAST result, and gene map (Supplementary Figs S2 and S3).
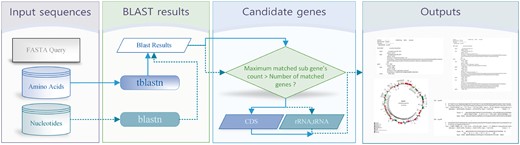
Overview of the AGORA system. There are four steps for annotating genes. The solid line indicates the flow of amino acids and the dotted line indicates the flow of nucleotides. Each line produces results that are finally provided as outputs
The second service module is implemented by Bootstrap and is designed for the user-friendly interface. Based on the IP address, the system creates a unique ID and stores it on the session at the browser. The web-based service provider manages multiple jobs using the information provided during the browser session. Therefore, the web server can control different jobs simultaneously.
3 Evaluation
The currently available tools for organellar genome annotation are DOGMA, GeSeq and CpGAVAS. The system specifications for the evaluation of AGORA are shown in Supplementary Table S1. For the organellar annotation, the user can select the references. Both DOGMA and CpGAVAS only use a restricted reference type, which is system-defined. However, GeSeq and AGORA can also upload the user-defined references. In terms of dealing with the genome type, all the three tools operate well for chloroplast genomes. However, the mitochondrial genome is not properly annotated, as shown in the Supplementary Tables S1 and S2. Another important issue for annotation is generating the GenBank file, because this file can be directly uploaded to NCBI if new genome sequences are annotated. Both GeSeq and AGORA support the GenBank file format, and CpGAVAS provides the GFF3 format. The GenBank file can be used to draw the circular map using OGDRAW tool, which is useful for quick analysis of the genome.
4 Conclusions
AGORA is a web-based organellar gene annotator, which uses reference sequence similarity. This tool can independently select the references, because the system supports both NCBI RefSeq by GenBank accession number and user-defined uploaded FASTA format reference sequences. By setting the user-defined option values, the user can identify the best matched gene or several candidate genes. Especially, if the option value is set to 1, the system provides a perfectly circular matched genome for not only the mitochondrion but also the chloroplast. Moreover, AGORA covers various species to annotate the organellar genome. The currently available tools are usually compatible only with plants, but AGORA annotates the mitochondrial genome of both plants and animals.
Funding
This research was supported by the National Research Foundation (NRF) of Korea funded by the Ministry of Science, ICT & Future Planning, Basic Science Research Program [MSIP; NRF-2016R1C1B1007929] to J.J; the Ministry of Education [2015R1D1A1A01057899] to J.I.K.; [2016R1D1A1A09919318] to G.Y.; This work was also supported by 2017 Hongik University Research Fund.
Conflict of Interest: none declared.
References


{kind=link}