





Abstract
ATAC-seq is rapidly emerging as one of the major experimental approaches to probe chromatin accessibility genome-wide. Here, we present ‘esATAC’, a highly integrated easy-to-use R/Bioconductor package, for systematic ATAC-seq data analysis. It covers essential steps for full analyzing procedure, including raw data processing, quality control and downstream statistical analysis such as peak calling, enrichment analysis and transcription factor footprinting. esATAC supports one command line execution for preset pipelines and provides flexible interfaces for building customized pipelines.
esATAC package is open source under the GPL-3.0 license. It is implemented in R and C++. Source code and binaries for Linux, MAC OS X and Windows are available through Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/esATAC.html).
Supplementary data are available at Bioinformatics online.
1 Introduction
Assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq) is a sensitive method to probe chromatin accessibility genome-wide (Buenrostro et al., 2013). The library preparation is fast, easy-to-perform and requires low amount of biological sample. These advantages make ATAC-seq become a popular way to study open chromatin, nucleosome positioning and transcription factor (TF) footprinting in cell lines or primary tissues by a booming number of laboratories.
Compared with its easy-to-perform experiment, ATAC-seq data analysis may take much more time and effort. Highly integrated cross-platform software to process ATAC-seq data is still lacking. Researchers need to set up their own local pipeline and use multiple tools, each of them provides partial functions of the entire data analysis workflow. Installing those tools from diverse sources, learning their manuals, testing their functions and integrating them together are tedious and time-consuming.
To fill this gap, we developed an easy-to-use R/Bioconductor package named ‘esATAC’. esATAC systematically integrates the state-of-the-art software for full procedure ATAC-seq data analysis, covering raw data processing, downstream statistical analysis and multiple quality control (QC) functions. For the ease of user, esATAC provides preset pipelines that can be executed by one command line under R/Bioconductor environment on different platforms. Advanced users can easily create customized pipelines through flexible interfaces in esATAC. Multi-core and memory control mechanisms have been implemented to optimize hardware utilization.
2 Design and implementation
The flowchart of esATAC is shown in Figure 1a.
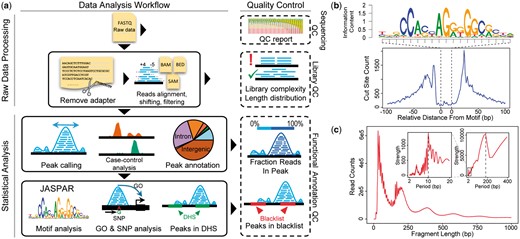
(a) esATAC workflow. esATAC pipeline is mainly divided into two parts, raw data processing and statistical analysis. QC functions at multiple levels are provided, including sequencing QC, library QC and functional annotation QC. (b) and (c) Examples of analyzing ATAC-seq data (GEO accession number GSE47753, see Supplementary Material). (b) CTCF footprinting. (c) Fragment length distribution. Periodicity of approximately 200 base pairs (bp) for nucleosome protection and 10.4 bp for the pitch of the DNA helix is shown by fast Fourier transformation in the upper right corner
2.1 Data analysis workflow
The workflow can be mainly divided into two parts, raw data processing and statistical analysis.
In the raw data processing part, esATAC can directly handle ATAC-seq raw data in FASTQ format. It wraps AdapterRemoval (Schubert et al., 2016) for adapter trimming and Bowtie2 (Langmead et al., 2012) for reads alignment. esATAC will sort the mapped reads, remove duplicates, shift reads for Tn5 insertion (Buenrostro et al., 2013) and generate intensity profile in BigWig format for genome browser visualization.
In the statistical analysis part, esATAC provides a comprehensive analyzing procedure for mapped ATAC-seq reads. It identifies open chromatin peak regions using F-seq (Boyle et al., 2008), which specializes in seeking genome-wide profiling of open chromatin regions with high sensitivity (Koohy et al., 2014). The peaks are annotated and related gene ontology terms are reported (see Supplementary Material). esATAC has integrated known TF motifs in JASPAR database (Mathelier et al., 2016) to find potential TF binding sites in the peak regions, and generate TF footprinting plots (Fig. 1b).
2.2 Quality control
esATAC provides multiple level QC functions. Raw sequencing reads quality report will be generated (Gaidatzis et al., 2015). esATAC performs fragment length QC analysis, providing that typical ATAC-seq fragment length distribution has a clear periodicity caused by nucleosome protection and the pitch of the DNA helix (Fig. 1c). Other QC methods adopted by ENCODE consortium have been integrated (see Supplementary Material), and concordance between replicates can be reported.
2.3 Implementation
For user convenience, we preset pipelines to analyze single sample and case-control paired samples for human and mouse. Users only need to provide the raw sequencing files and can execute the entire pipeline with one command in R. Dependent data like annotation files and bowtie2 index can be downloaded and built automatically. An HTML summary report for comprehensive QC and statistical analysis will be generated.
The package is managed by dataflow graph, therefore users can easily understand and trace the pipeline processing modules (see Supplementary Material). Mechanisms in esATAC such as inputs legality checking ensure that sophisticated users are able to customize the pipeline or integrate other tools from any intermediate stages easily.esATAC provides memory control and parallel computing options to maximize the computing efficiency. Breakpoint detection has been established to ensure that users do not have to redo the finished processes in case the program was interrupted.
3 Conclusion
We proposed esATAC aiming to make ATAC-seq data analysis easy for a wide range of users. esATAC covers whole procedure for ATAC-seq data processing. It can be installed on different platforms and perform ‘one command line for result’ analysis. Users without sophisticated programming skills can get started easily. At the same time, all the sub-functions are componentized, making it a flexible platform for advanced users to build pipelines for specialized applications.
Funding
This work was supported by the National Science Foundation of China [grant nos. 31371341, 61773230 and 61721003], Tsinghua University Initiative Scientific Research Program [no. 20141081175] and the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University.
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Zheng Wei and Wei Zhang authors should be regarded as Joint First Authors.


{kind=link}