





Abstract
Genetic reporter assays are a convenient, relatively inexpensive method for studying the regulation of gene expression. Massively Parallel Reporter Assays (MPRA) are high-throughput functionalization assays that interrogate the transcriptional activity of many genetic variants at once using a library of synthetic barcoded constructs. Despite growing interest in this area, there are few computational tools to design and execute MPRA studies.
We designed an online web-tool and R package that allows for interactive MPRA experimental design encompassing both power analysis and design of constructs. Our tool is tuned using data from real MPRA studies. Users can adjust experimental parameters to examine the predicted effect on assay power as well as upload VCFs for automated construct sequence generation.
The MPRA Design Tools web application is available here: https://andrewghazi.shinyapps.io/designmpra/, https://github.com/andrewGhazi/designMPRA and https://github.com/andrewGhazi/mpradesigntools.
Supplementary data are available at Bioinformatics online.
1 Introduction
Many factors can reduce the sensitivity of MPRAs. Variants prioritized by GWAS association signal may represent functional classes that cannot operate in an episomal setting such as MPRA; for instance variants that only function in certain cell types or conditions may be undetectable by MPRA. Moreover, variants tested by MPRA may be non-functional yet emerge as testable candidates from GWAS due to linkage disequilbrium. In addition to these and potentially other factors, noise within the assay reduces the statistical power to detect variant function because the mRNA output per DNA input can be highly variable. Our analysis of published MPRA studies (Tewhey et al., 2016; Ulirsch et al., 2016) has shown that the standard deviation of activity across barcoded replicates of a given oligonucleotide is on average around .95, corresponding to a 2.6-fold difference in mRNA per input DNA molecule (see Supplementary Data S2). This level of noise suggests the assay cannot sensitively detect functional variants with small effect sizes. This can be addressed in part by increasing the number of barcodes per construct as well as through repeated transfections.
Here we show MPRA Design Tools for interactive design of MPRA studies. Users can adjust parameters such as barcodes per allele and activity variance to examine the estimated effect on statistical power, as shown by a synthetic demonstrative example in Figure 1. After selecting parameters, a tab allows users to upload VCFs of their variants to obtain MPRA construct sequences based on the hg38 genome. A companion R package provides more customizable sequence generation features.
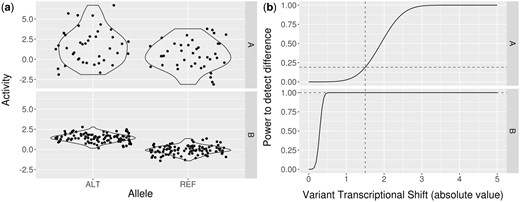
(a) Two synthetic example MPRA outputs for hypothetical variants (see Supplementary Data S2) in scenarios A and B with transcriptional shifts of 1.5. Variant A uses 40 barcodes per allele and exhibits a standard deviation of 2 while B uses 100 barcodes per allele and exhibits a standard deviation of .5. Variances are based on observed data from Ulirsch et al. (2016) and Tewhey et al. (2016). (b) Line plots showing power to detect changes using the given multiplicities of barcodes and standard deviations in a MPRA assay with 1000 variants. In scenario A the statistical power to detect the shift is less than 20% while in scenario B the power is effectively 1. This example shows the necessity of selecting design parameters that maximize experimental efficiency
Our tool differs from existing software such as MPRAnator (Georgakopoulos-Soares et al., 2016) in both ease-of-use and interactivity. The MPRA Design Tools guides the user to choose experimental parameters that best fit goals while the sequence generation can be done either with VCF upload or using the R package. Our tool acquires genomic context from the hg38 reference genome, rather than requiring input by the user.
2 MPRA statistical power
Variant activity measurements are noisy; therefore, a set of multiple barcodes are used for each allele. These activity measurements are approximately normally distributed (see Supplementary Data S3), so our tool uses this assumption and published data (Tewhey et al., 2016; Ulirsch et al., 2016; see Supplementary Data S2 and S3) to estimate statistical power of detecting true shifts at a range of effect sizes (see ‘Power’ tab; see Supplementary Data S4 for modelling details). Changes to input parameters automatically update the plot output. This interface allows users to design experiments that optimize statistical power.
3 MPRA sequence generation
MPRA experiments require thousands of uniquely barcoded sequences that need to meet specific design parameters. The barcodes must be unique, must not generate restriction sites that would cause the constructs to be degraded in the plasmid library, and must be otherwise transcriptionally inert (see Supplementary Data S5). Additional parameters such as sequence context range or restriction enzymes may be adjusted.
We added functionality to allow for automatic generation of MPRA sequences based on variants input by the user. After inputting the number of barcodes per allele, the range of sequence context and other design parameters, users provide a VCF containing variants to receive a tab-separated file containing the MPRA sequences for their experiment.
4 Conclusions
MPRA Design Tools allows users to rapidly and interactively design MPRA experiments. The tool is available by web and R source.
Funding
This study was supported by United States National Institutes of Health Grant R01HL128234.
Conflict of Interest: none declared.
References


{kind=link}