





Abstract
With the growing availability of population-scale whole-exome and whole-genome sequencing, demand for reproducible, scalable variant analysis has spread within genomic research communities. To address this need, we introduce the Python package Variant Analysis and Prioritization (VAPr). VAPr leverages existing annotation tools ANNOVAR and MyVariant.info with MongoDB-based flexible storage and filtering functionality. It offers biologists and bioinformatics generalists easy-to-use and scalable analysis and prioritization of genomic variants from large cohort studies.
VAPr is developed in Python and is available for free use and extension under the MIT License. An install package is available on PyPi at https://pypi.python.org/pypi/VAPr, while source code and extensive documentation are on GitHub at https://github.com/ucsd-ccbb/VAPr.
1 Introduction
Precision medicine’s promise of diagnoses and treatments precisely tuned to each patient cannot be realized without a comprehensive understanding of the genomic variants underlying individual differences (Morgan et al., 2010; Lek et al., 2016). The rise of next-generation sequencing has drastically decreased the cost and increased the availability of the raw genomic data upon which such understanding is built, but the technical hurdles to mining full annotation of these data, especially for variants from population-scale whole genome sequencing, hinder the transformation of variant information into knowledge (Pabinger et al., 2014).
Due to the glut of sequencing data and the scarcity of computational biologists with deep specialization in variant analysis, much variant annotation and prioritization must be performed by computer-savvy lab biologists and by bioinformatics generalists. Currently, they are hampered by the necessity of learning details of multiple annotation software tools and integrating these into an analysis environment, as well as developing bespoke data structures supporting storage and filtering of variants’ extremely heterogeneous information (Beck et al., 2014). Given these issues, many such analyses are performed manually or semi-manually, using ad hoc filtering approaches that are difficult to accurately record and reproduce. Variant Analysis and Prioritization (VAPr) addresses these challenges by providing a simple software package that painlessly integrates high-quality reproducible variant investigation into any analysis pipeline.
2 Main features
The VAPr workflow is described in Figure 1. As a wide variety of tools already exist for annotating variants (Pabinger et al., 2014), VAPr builds upon the best-in-class choices from this field. For basic evaluation of novel variants, VAPr leverages the popular command-line software ANNOVAR (Wang et al., 2010); since this tool requires the installation and update of various local data files, VAPr also provides set-up methods to easily manage these resources. For collation of existing annotations for known variants, VAPr queries the MyVariant.info web service (Xin et al., 2016), which aggregates continually updated information from more than a dozen disparate variant annotation efforts. Both sets of resulting annotations for each variant are merged and recorded in a local NoSQL MongoDB, against which filtering and output steps are performed. The BigQ extension to the i2b2 framework for clinical research management (Gabetta et al., 2015) has already demonstrated that such databases provide fast, scalable and flexible storage for variant information, and VAPr thus extends these benefits to the large body of data analysts working outside the i2b2 framework. Access to all data is provided by a single Python API (Application Programming Interface); furthermore, since VAPr is available through PyPi, it can be installed with a simple one-line command (pip install VAPr).
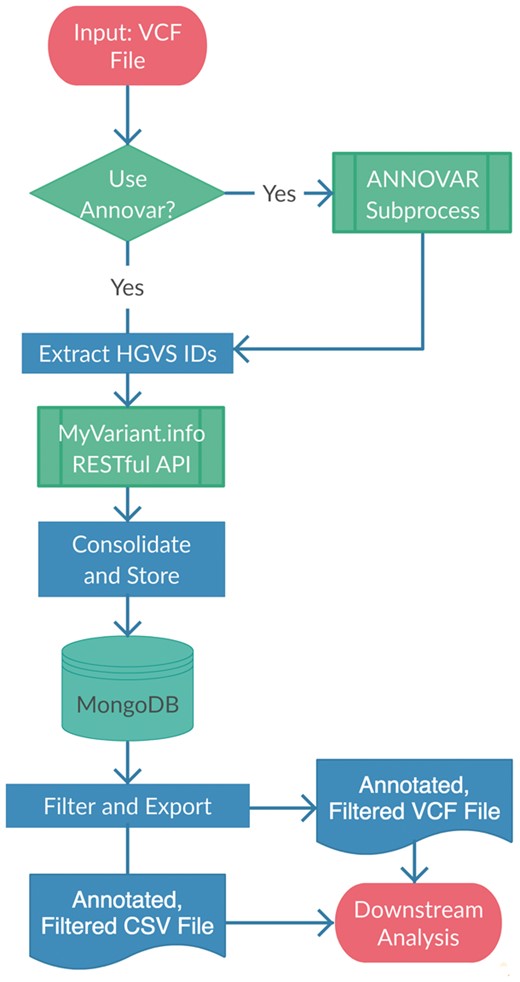
VAPr workflow for variant annotation and prioritization. Variant calls in VCF format are parsed, annotated, aggregated and stored in a MongoDB database where they can be quickly retrieved through filtering queries or output as CSV files
An additional advantage of VAPr’s storage approach over a traditional relational database is that users need not learn a complex table structure in order to filter for variants of interest (Schulz et al., 2016). VAPr manages the data in such a way that custom queries can be formulated using only MyVariant.info or ANNOVAR field names using the standard PyMongo syntax (O’Higgins, 2011). Many users, however, will find that they do not need to write custom queries at all, as VAPr provides pre-built variant analysis filters for four classes of variants that are commonly of interest: deleterious rare variants, known disease variants, deleterious compound heterozygous variants, and de novo variants from trios. The full definitions for these filters are detailed in the VAPr documentation. A user requiring different filtering criteria can easily create a custom query using these preset filters as a template.
As variant data are primarily conveyed via the variant call format (VCF) (Danecek et al., 2011), VAPr supports single-sample and multi-sample VCF file input and output. VAPr results can also be exported to comma-separated value (CSV) files for examination in an Excel spreadsheet and/or integration with a wide variety of downstream analysis tools. VAPr supports human genome assemblies hg19 and hg38 and comes with extensive user documentation that presupposes no expertise beyond basic experience with Python, including a tutorial on how to compose custom queries. In addition, we provide installation instructions, an extensive suite of unit tests and a fully executable Jupyter Notebook workflow to demonstrate a sample usage of VAPr.
3 Sample implementation
VAPr supports extremely lightweight implementation of variant investigation in Python scripts. The code sample below demonstrates the minimal code necessary to read in and parse a VCF file, annotate its variants with MyVariant.info and ANNOVAR and identify those meeting criteria for known disease variants. As shown here, these tasks can be accomplished with only four statements.
from VAPr import vapr_core.VaprAnnotator
# Parse, annotate, and save variants from input VCF
annotator = VaprAnnotator(“/path/to/vcfs/”, “/path/to/output_dir/”, mongo_db_name, mongo_ collection_name, “/path/to/annovar/”, build_ver=“hg19”)
dataset = annotator.annotate()
# Identify and prioritize known variants
known_disease_variants = dataset.get_known_disease_variants()
Run times on a laptop computer with four cores and 16GB memory were 2.5 min for a whole exome vcf file (40 000 variants) and 5 h for a whole genome vcf file (4.8 million variants).
4 Conclusions
VAPr provides a simple, turn-key Python API that integrates popular annotation tools with a flexible local data store to efficiently annotate, analyze and prioritize population-scale variant calls. It accommodates large variant files (such as those from whole genome sequencing) and meshes easily with upstream variant calling pipelines, while offering simplified Python-based custom filtering for variant prioritization and built-in filter options for common criteria. Furthermore, as VAPr is entirely open-source and available for expansion under the MIT license, users can easily extend it to include their own prioritization strategies and thus reduce dependence on manual variant filtering. We hope that, given its native ease-of-use as well as its extensibility, VAPr will empower lab biologists and non-specialist bioinformaticists to perform more transparent and reproducible analyses. Such reproducible, automated variant evaluation methods are critical not only for primary research but also in realizing many precision medicine workflows.
Funding
This work was supported by the National Institutes of Health [UL1TR001442] of CTSA and the San Diego Center for Systems Biology [P50GM085764]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Amanda Birmingham, Adam M. Mark and Carlo Mazzaferro authors should be regarded as Joint First Authors.


{kind=link}