





Abstract
Software pipelines have become almost standardized tools for microbiome analysis. Currently many pipelines are available, often sharing some of the same algorithms as stages. This is largely because each pipeline has its own source language and file formats, making it typically more economical to reinvent the wheel than to learn and interface to an existing package. We present Plugin-Based Microbiome Analysis (PluMA), which addresses this problem by providing a lightweight back end that can be infinitely extended using dynamically loaded plugin extensions. These can be written in one of many compiled or scripting languages. With PluMA and its online plugin pool, algorithm designers can easily plug-and-play existing pipeline stages with no knowledge of their underlying implementation, allowing them to efficiently test a new algorithm alongside these stages or combine them in a new and creative way.
We demonstrate the usefulness of PluMA through an example pipeline (P-M16S) that expands an obesity study involving gut microbiome samples from the mouse, by integrating multiple plugins using a variety of source languages and file formats, and producing new results.
Links to github repositories for the PluMA source code and P-M16S, in addition to the plugin pool are available from the Bioinformatics Research Group (BioRG) at: http://biorg.cis.fiu.edu/pluma.
1 Introduction
A pipeline is a software design strategy that is applicable to software packages within and outside of bioinformatics. The idea is to assemble a software execution using sequential modules or stages, with the output of a stage serving as input to one or more future stages. Stages are often developed independently, with file I/O serving as the bridge between adjacent stages. Software pipelines are particularly useful in -omics analysis because it is very likely that, independent of the type of -omics, there will at least be some distinct stages of processing raw data or intermediate results, followed by some downstream analysis to yield useful conclusions. Figure 1 shows a basic pipeline often used for metagenomics, with raw sequence data passing through a preprocessing stage that removes poor-quality reads, followed by a stage that groups or clusters reads by some similarity metric into operational taxonomic units (OTUs), and finally a stage that taxonomically classifies these OTUs using database queries into the closest taxon match within a phylogenetic tree.

A basic metagenomics pipeline. Raw sequence reads are filtered for quality during a denoising step, followed by clustering into groups (called OTUs) and a labeling of each OTU with the closest corresponding phylogentic classification
At this point, many software pipelines have been assembled for microbiome analysis. This is especially true now that multi-omics approaches (Segata et al., 2014) are being used. Currently, Biostars (Parnell et al., 2011) contains references to forty pipelines for metagenomics alone. A closer look at these pipelines shows that their differentiating factor is often one stage, and usually not the entire metagenomics analysis. For example, TETRA (Teeling et al., 2004) and MEGAN (Huson et al., 2011) differ in the clustering stage; the first clusters based on composition and the latter based on similarity. In cases where the overarching purposes are the same—i.e. Qiime (Caporaso et al., 2010) and Mothur (Schloss et al., 2009) are both microbial diversity analysis tools, their implementation differs. Mothur offers C++ functionality that is transparently invoked through domain-specific language (DSL) commands, while Qiime implements its stages as scripts.
This explosion in analysis pipelines with little variation between them demonstrates that a significant amount of reinventing the wheel is currently taking place, slowing down the development and dissemination of new and useful algorithms. Although all stages of a pipeline other than a newly developed one may currently exist elsewhere, integrating this new stage often requires (i) basic knowledge of another pipeline including its input formats and how to run it, (ii) some knowledge of its underlying software design and languages and/or (iii) a heavy installation process. These issues become more acute when a developer wants to combine multiple stages from multiple pipelines to produce something new. Considering the time this may require, it is conceivably faster to simply reconstruct these stages in a language with which they are more familiar, resulting in yet another pipeline.
This issue has not gone unnoticed in the bioinformatics field. The National Institute of Health (NIH) developed Nephele (Battré et al., 2010) as a centralized portal of publicly available pipelines for microbiome analysis. Their strategy is to try to anticipate all commonly used pipelines and file formats and provide a ‘pipeline pool’. Their site already illustrates how quickly these pipelines can explode, i.e. they have six different pipelines that perform 16S analysis with Qiime, that use six different file formats. Their targetted audience, data analyzers, differs from ours. Algorithm developers will still have the same issues with Nephele because their pipelines are built with existing tools, mainly Qiime and Mothur. Looking at these two packages, Qiime has traditionally offered flexibility through scripting. An algorithm developer could design their pipeline stage as a script (or program invoked by a script) and insert it alongside other Qiime scripts, forming a complete pipeline. While flexible, this design still requires compatibility with Qiime file formats, and also some underlying knowledge of Qiime scripts so that the developer can invoke them appropriately. Mothur removes this latter issue by performing stage invocation through a higher level domain-specific language (DSL) but still requires compatibility with their file formats, and in addition any new pipeline stage would need to be callable from the Mothur DSL, requiring some C/C++ interfacing. BPipe (Sadedin et al., 2012) and NextFlow (Di Tommaso et al., 2017) provide bridges between scripted stages that are heterogeneous with respect to programming language, but still require knowledge of how to run each script and their file formats because the user must specify the command(s) to execute each stage. Snakemake (Koster and Rahmann, 2012) performs a similar task by defining a pipeline as a sequence of rules, but each rule requires the command for running its respective package, creating a similar issue. This leads us to our first desirable quality for the type of framework we envision: for an algorithm developer to channel all of their energy into developing and testing their algorithm, there should ideally be zero interfacing to an existing computational core required. All of these standalone pipelines that we mention each require some form of interfacing to their software, through code or input formats (or both), as we show in Table 2. Note that language flexibility and lightweightness (our other two desirable qualities, see Table 1) are hit or miss with these pipelines depending on design; for example Qiime is more than ten times the size of Mothur but users can develop in their language of choice since everything is scripted. Mothur requires you to use their DSL or build C++ libraries for interfacing so this language flexibility is taken away, although their software is more lightweight. As we will see, this tradeoff extends even to other categories of pipelines that succeed at achieving zero interfacing.
The three characteristics that we envision as most important for facilitating algorithm development and testing, and the question(s) they attempt to answer
| Quality | Question |
|---|---|
| Zero Interfacing | Can I avoid learning an existing platform? |
| Language Flexibility | Can I develop in my language of choice? |
| Lightweightness | Can I test/debug with minimal resources? |
| Quality | Question |
|---|---|
| Zero Interfacing | Can I avoid learning an existing platform? |
| Language Flexibility | Can I develop in my language of choice? |
| Lightweightness | Can I test/debug with minimal resources? |
The three characteristics that we envision as most important for facilitating algorithm development and testing, and the question(s) they attempt to answer
| Quality | Question |
|---|---|
| Zero Interfacing | Can I avoid learning an existing platform? |
| Language Flexibility | Can I develop in my language of choice? |
| Lightweightness | Can I test/debug with minimal resources? |
| Quality | Question |
|---|---|
| Zero Interfacing | Can I avoid learning an existing platform? |
| Language Flexibility | Can I develop in my language of choice? |
| Lightweightness | Can I test/debug with minimal resources? |
Comparing and contrasting the three popular types of analysis pipelines, and how they facilitate what we envision as the three most important qualities when facilitating algorithm development and testing
| Type | Zero Interfacing | Language flexibility | Lightweight |
|---|---|---|---|
| Standalone (Qiime, Mothur) | No | Depends | Depends |
| Workflows (Taverna, KNIME) | Yes | No | Depends |
| Superpipelines (Galaxy, Docker) | Yes | Yes | No |
| Type | Zero Interfacing | Language flexibility | Lightweight |
|---|---|---|---|
| Standalone (Qiime, Mothur) | No | Depends | Depends |
| Workflows (Taverna, KNIME) | Yes | No | Depends |
| Superpipelines (Galaxy, Docker) | Yes | Yes | No |
Comparing and contrasting the three popular types of analysis pipelines, and how they facilitate what we envision as the three most important qualities when facilitating algorithm development and testing
| Type | Zero Interfacing | Language flexibility | Lightweight |
|---|---|---|---|
| Standalone (Qiime, Mothur) | No | Depends | Depends |
| Workflows (Taverna, KNIME) | Yes | No | Depends |
| Superpipelines (Galaxy, Docker) | Yes | Yes | No |
| Type | Zero Interfacing | Language flexibility | Lightweight |
|---|---|---|---|
| Standalone (Qiime, Mothur) | No | Depends | Depends |
| Workflows (Taverna, KNIME) | Yes | No | Depends |
| Superpipelines (Galaxy, Docker) | Yes | Yes | No |
One possibility to gain zero interfacing is through more general workflow engines like Taverna (Wolstencroft et al., 2013) and KNIME (Berthold et al., 2007). They offer an interactive development environment (IDE) where developers can both construct new algorithms as components and establish workflows with components as stages. However, their component development language is limited to Java. Kepler (Altintas et al., 2004) similarly restricts development of new extensions to R, and Ruffus (Goodstadt, 2010) can handle only Python extensions. This can be a cost of a homogeneous development and user environment, since IDEs often are catered to a specific programming language. Therefore we observe that while workflow engines achieve minimal to zero interfacing a user must use the programming language supported by their engine, taking away the desirable quality of language flexibility as we illustrate in Table 2. Language flexibility, or the ability to prototype in a programming language of choice, allows a developer to construct an algorithm with no additional overhead in learning a new syntax for expressing their thoughts. Lightweightness with workflows will once again depend on design.
The alternative to attaining zero interfacing is to anticipate all possible user needs, and encapsulate a union of all these requirements into the package itself (we refer to these as superpipelines). This was the idea behind Dockerized containers (Merkel, 2014), which can be chained together to form pipelines (Narayanasamy et al., 2016). Superpipelines by definition facilitate language flexibility, since multiple language tools can be included. These can be chained together and sequentially executed. However, the additional requirements for the runtime environment (which includes system tools in addition to language tools) will likely increase the weight of these containers to well beyond the algorithm itself. Also, some of these additional requirements will likely be replicated (i.e. two containers built for the same system with the same dependencies), adding unnecessary additional weight. Galaxy takes a slightly different approach, providing flexibility through a generalized interface by which a user can insert complete software packages as pipeline stages. To maximize generality, their software includes accessibility to multiple large-scale online databases, allowing the user to choose the best option for their work. Their package also interacts with a large number of existing software packages (more than forty), once again improving its generality. The downside to this is that the installation is not lightweight, requiring more than 40 additional packages and hundreds of MB of storage. Thus in general, we observe with superpipelines a general loss of our third desirable quality of lightweightness, since almost by definition these will try to encapsulate a large set of possible user requirements to maximize flexibility. We illustrate this in the final row of Table 2. Lightweightness is desirable particularly in algorithm development and test stages where small datasets are typically used, because a heavy installation process with many large packages may not be practical for an algorithm developer and their often limited resources, and certainly not desirable if test datasets can be stored locally.
Our goal is to make algorithm development, testing and debugging as easy as possible. First, we remove any requirement of interfacing to an existing back end (zero interfacing), allowing new extensions to be individually compiled and dynamically loaded upon runtime reference in a user interface [configuration file or Graphical User Interface (GUI)]. We also provide a uniform interface for all extensions, removing the requirement of interfacing to additional extensions in the same pipeline. File format conversion plugins can be added as necessary to pipelines with no loss of generality. If a developer wants to test a new algorithm and it follows a pipeline stage that outputs an incompatible file, they could add a new stage before their algorithm with the appropriate file converter. In our tool, Plugin-Based Microbiome Analysis (PluMA), these dynamically loaded stages are called plugins (Alexandrescu, 2001). Many plugins have already been packaged and/or developed and can be individually downloaded from our online plugin pool. It should be noted that the newest release of Qiime (version 2) also offers plugin compatibility, but their plugin source language is limited to Python. Our final goal runs alongside Mothur, in that we minimize any need to understand plugin implementation details. An algorithm developer should be able to construct plugins in a language in which they are fluent and/or that makes the most sense given their requirements, and seamlessly run them alongside plugins written in languages that they may not know. PluMA thus supports a range of compiled and scripting languages for plugin implementation with an interface that remains uniform independent of their source language, achieving language flexibility. Finally, because plugins are dynamically loaded a user can simply run PluMA’s small computational core (182 KB) with only the plugins they need and count on them loading properly at runtime, creating a lightweight environment for development and testing. Our ultimate goal with PluMA is to nurture and grow communities that make use of analysis pipelines, through a transparent and simple interface that removes reinventing of the wheel (Prlic and Procter, 2012).
The flexible and lightweight design of PluMA also offers additional benefits. With our Plugin Generator (PluGen), existing or newly developed software tools can be converted to plugins and combined in new and unique ways to suit the needs of a specific dataset. PluGen allows a developer to seamlessly integrate useful standalone tools such as Qiime and Mothur into a lightweight and flexible pipeline alongside other plugins (i.e. their algorithm). Though lightweightness results in some limits when the plugin pool is small, our philosophy is to provide enough incentive to use PluMA, then count on the user base itself to grow the plugin pool to levels that meet their needs. The greatest advantage to this strategy as a long-term investment is that these features will become plugins with uniform interfaces that are executable in a lightweight environment and thus easily insertable into other users’ pipelines, attacking the problem more at the root by facilitating usability. We illustrate PluMA through an example metagenomics pipeline (P-M16S) that analyzes 16S gut microbiome data from the mouse, and finally we will show the results of P-M16S, their implications and our vision for the future of PluMA.
2 System and methods
The core software design pattern that makes PluMA extensible and lightweight is the Plugin (Alexandrescu, 2001). We summarize our application of this pattern in Figure 2. The idea is to create a small and for all intents and purposes infinitely extensible executable file. A simple back end (in this case PluMA) has the sole responsibility of dynamically loading and executing plugins. New pipeline stages become plugins, which are always compiled (if necessary) as standalone modules and dynamically loaded. The choice of which plugins to load is governed by a runtime user selection through an interface (in this case through a configuration file, though a GUI could easily be substituted). For our example, only plugins A and C would be loaded, though plugin B could easily be loaded or substituted by changing the user input.
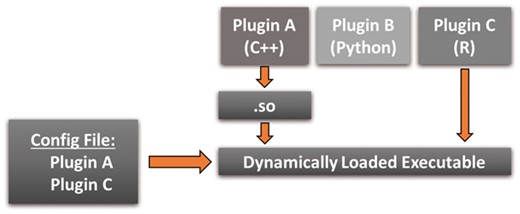
The Plugin design pattern
Using this pattern offers several advantages. The executable, which resides on permanent storage, is very lightweight and involves only PluMA itself. The runtime executable is also lightweight, only including PluMA plus the exact plugins necessary for the pipeline. This advantage extends to source code as well; for example, even though PluMA could run plugin B, there is no requirement to install the code for B if a user will never run a pipeline with B. Most importantly, extending the package becomes easy because an algorithm developer can just focus on their individual plugin and not worry about how to interface it to PluMA. As long as they design their plugin using a compatible Application Programming Interface (API, which we will see is straightforward), they can set up pipelines with their new algorithm through the user interface.
2.1 Heterogeneous plugins
With the source code of PluMA in C++ we can run plugins in C/C++ for the CPU or CUDA (Nickolls et al., 2008) for the Graphics Processing Unit (GPU). These would be compiled into shared libraries, similar to plugin A in the figure. We also allow plugins in (at the moment) Python, Perl and R—all of which have interpreters that use this same C runtime environment, becoming compatible through various interfaces (Eddelbuettel, 2013; Quillan, 1998; van Rossum, 2005). Algorithm developers thus have great freedom of choice regarding their source language, and through the user interface can test alongside plugins built using different languages.
2.2 Plugin pool
This methodology enables us to create a public plugin pool of available plugins written in various programming languages. Our current plugin pool contains sixty-five plugins, which we hope will continue to grow with time via a community effort. To execute PluMA, a developer can just download PluMA and the plugins they require to assemble a pipeline that tests their new algorithm. Once completed they can e-mail us the plugin and we will add it to the plugin pool. As the PluMA user base grows and pipelines are assembled we immediately expect a growth in file converters for the most popular formats (indeed about 10% of the pool currently consists of converter plugins), and that overtime it will become highly likely that almost all necessary file converters will be available as plugins. This carries an added bonus that they will also be accessible within PluMA’s easy plug-and-play environment, which removes obligation for finding one themselves that could be written in any language and use any interface. When developing future PluMA releases, we will be sure to rerun all plugins in the plugin pool to ensure compatibility. We thus envision the plugin pool of PluMA as an ultimate portal from which users can download pipeline stages and assemble different types of microbiome analysis pipelines in an efficient and easy-to-use manner for testing and debugging. Similar portals can be created for other bioinformatic analysis pipelines.
2.3 Experimental pipeline: P-M16S
We illustrate the flexibility of PluMA by using it to construct a pipeline (P-M16S) that expands the study of (Kozich et al., 2013). They studied the connections between the mouse gut microbiome and obesity by placing mice on a specific diet and taking microbiome samples the first ten days (when they were not yet obese), and the final ten days (days 141–150 of their new diet), after weight gain. Their analysis produced interesting results, showing differences in β-diversity before and after weight gain, and also correlating weight gain with an increase in the abundance of the Porphyromonadaceae microbial family. They used the SILVA (Quast et al., 2013) bacterial reference to align sequences; for classification, they used a Bayesian classifier in Mothur with a properly formatted training set from the Ribosomal Database Project [RDP (Larsen et al., 1993)].
P-M16S will analyze the relationships between bacterial OTUs before and after weight gain. Microbiomes are highly complex because of ecological relationships (i.e. cooperation, competition, etc.) between the entities (Costello et al., 2009), and studying these relationships can help deepen this knowledge. Bacterial co-occurrence networks (Faust et al., 2012) offer one option, as they provide an idea of which bacteria tend to occur together and which tend to avoid each other. P-M16S will apply centrality (Easley and Kleinberg, 2010) to these networks to infer the most important bacterial taxa in this network of relationships (Dempsey and Ali, 2011). The assumption is that the most critical relationships may be between taxa that are not necessarily the most abundant.
Figure 3 shows our P-M16S pipeline as a series of PluMA plugins and their source languages. Since we are performing analysis before and after weight gain we will run this same pipeline twice, once for the first ten samples (we call this Early) and once for the last ten samples (we call this Late). Mothur and Cytoscape (Shannon et al., 2003) have been installed on our machine, and we use our Plugin Generator (PluGen) to produce a C++ plugin for each. The first portion of the experiment (up to the step of OTU classification) uses the Mothur plugin and as shown in (Kozich et al., 2013) produces a set of OTUs and normalized abundances.

Our test metagenomics pipeline, P-M16S. Note plugins have been implemented in various programming languages, and some have been generated by PluGen. File conversion plugins have been downloaded from the PluMA plugin pool
The language R offers many useful libraries for computing correlations between OTUs (including Pearson, Spearman, Kendall, etc.) We thus build this plugin with R, as stage four of our pipeline. We use a file conversion plugin called CountTableProcessing (written in R) from the PluMA plugin pool to convert the output of Mothur to Comma-Separated-Value (CSV) format. Another plugin called CSVNormalize converts the file to normalized form for further processing.
For computing centrality we will use the algorithm Ablatio Triadum [ATria (Cickovski et al., 2017)] which operates on signed and weighted networks. Note that correlation networks are signed and weighted. ATria has been shown to find important nodes (positive and negative) in spatially different regions of the network (including leaders of subnetworks or clubs, common enemies or villains, and bridge nodes that connect nodes from multiple clubs). We implement a plugin for ATria using CUDA, which runs efficiently by taking advantage of massive parallelism offered by the GPU. The ATria plugin is preceded by another file conversion plugin from the plugin pool (written in Python), which pads the CSV file output by R (using write.table).
Cytoscape is a useful tool for network visualization. However, it does not support CSV format but does support the Graph Modeling Language [GML (Himsolt, 1995)]. We thus use one final file converter plugin from the pool, CSV2GML (also Python), to convert our network into a format that Cytoscape can visualize. The plugin for Cytoscape, like the plugin for Mothur, was generated by PluGen.
3 Implementation
We now describe how to add two new plugins, one in a scripting language (R) and another in a compiled language (CUDA). Once completed, we will seamlessly integrate them into P-M16S by supplying PluMA with the configuration file in Program 1. Note this particular example runs the Early samples, but Late would essentially look the same. We name the plugins Correlation and ATria, which must be unique. Note that each has an associated inputfile and outputfile, which can be none.
# Metagenomics analysis pipeline: Early
Prefix data/Early
Plugin Mothur inputfile Early.mothur outputfile none
Plugin CountTableProcessing inputfile Early.unique_ list outputfile abund.csv
Plugin CSVNormalize inputfile abund.csv outputfile abundN.csv
Plugin Correlation inputfile abundN.csv outputfile corr.csv
Plugin CSVPad inputfile corr.csv outputfile corrP.csv
Plugin ATria inputfile corrP.csv outputfile corrP.ATria.noa
Plugin CSV2GML inputfile corrP.csv outputfile corrP.gml
Plugin Cytoscape inputfile corrP.gml outputfile none
The generated Mothur plugin accepts Early.mothur, which contains almost the same set of Mothur commands used in (Kozich et al., 2013), except that it operates only on the Early (first ten) samples. The input of each stage gets generated by some previous stage (Early.mothur automatically produces a set of files that start with the prefix Early.unique_list). This need not be the immediately preceding stage (i.e. corrP.csv gets passed to both ATria and CSV2GML, because we need the correlation matrix to compute centrality, but also to visualize with Cytoscape after a conversion to GML). Plugins can be easily swapped in and out using comments (#), and the Prefix specifies a relative path for all input and output files (this can be done multiple times). Note that we freely specify these plugins with no knowledge of their underlying implementation, exposing the necessary parameters only (List et al., 2017). Furthermore, it is also clear that our techniques allow pipelines that go beyond just linear dependencies, and can implement a general acyclic network of dependencies.
3.1 Scripted plugin: Correlation
We now focus on the two missing stages. When adding new plugins, PluMA will automatically find and load them as long as we put them in a location specified by the environment variable PLUMA_PLUGIN_PATH (or in the source tree of PluMA). The only requirement applies to the functions that will be invoked by PluMA when executing this plugin: input(), run() and output(). These names are the same independent of the language of choice. input() and output() accept character string parameters: input() accepts the inputfile (which is automatically linked to its configuration file value) and output() analogously accepts the outputfile. We can put code anywhere, but this breakdown maintains a consistent interface and will generally be: input() reading the inputfile and populating internal structures, run() executing the algorithm and output() taking the internal structures and sending data to the outputfile. In this sense PluMA plugins achieve zero interfacing: no effort is required to interface to the computational core. The computational core instead interfaces to the plugins, by calling their functions.
libs <- c(“Hmisc”);
input <- function(inputfile) {
abund ≪- read.csv(inputfile, header = TRUE); }
run <- function() {
otus ≪- colnames(abund)
otus ≪- otus[2: length(otus)]
abundT ≪- t(apply(abund[,-1], 1, as.numeric));
result ≪- rcorr(abundT[,]);
corr ≪-as.matrix(result$r);
corr[which(result$P > 0.01)] ≪- 0; }
output <- function(outputfile) {
write.table(corr, file = outputfile, sep=“,”,
append = FALSE, row.names = unlist(otus),
col.names = unlist(otus), na=““);}
R correlation functions are included in the Hmisc library at the top of the file. Not all code must be in the three mentioned functions, some can be global or in other functions. Globally-scoped code in a scripted plugin is run when the plugin is loaded, and code in other functions will run if explicitly called within the plugin. In our case, input() reads a CSV file (inputfile) and populates the abundance matrix. run() then preprocesses this matrix, computes its transpose, and uses the rcorr method of Hmisc to compute the correlation matrix. Although rcorr defaults to Pearson correlations, others (i.e. Spearman or Kendall) can be easily passed, producing a matrix with values entirely within the range [–1, 1]. run also P-value thresholds this matrix. Finally, our output function sends this matrix to the specified outputfile, also assumed to be in CSV format. This simple example shows how an R plugin can be developed and integrated into a pipeline for testing using PluMA. Similar approaches would be taken for any R plugin, and as mentioned any general plugin would use the same three mentioned functions.
3.2 Compiled plugin: ATria
Since the source code for ATria is a bit involved and our goal is not to present that algorithm but to illustrate the features of PluMA, we use skeletons of our plugin code for ATria, written in CUDA to take advantage of the parallelism offered by GPUs. CUDA was developed by NVIDIA and runs on a graphics card using a shared memory architecture, allocatable to different threads and processors. The hardware is thus both highly multithreaded and highly multicore.
CUDA also extends C, and more recent releases of the NVIDIA compiler accept C++ syntax, which we also use here. Program 3 shows a template of our header file, with our new ATriaPlugin class extending the Plugin class of PluMA. We use the same input(), run() and output() methods as public member functions. Similar to the scripted plugin, we can declare any other functions or variables that we choose. In this particular case we use two GPU functions or kernels, written in CUDA. _GPU_Floyd includes an implementation of the Floyd-Warshall (Floyd, 1962) shortest path algorithm on the GPU, modified to use multiplication (since with signed and weighted networks with edge weights in the range [–1, 1] the longest magnitude path between two nodes will be the product (not sum) of its edges). The second, _GPU_Pay, will add largest-magnitude paths of gain and loss between a node and all other nodes to determine its ‘Pay’ or centrality. We have additional kernels as well, set up similarly.
#ifndef ATRIAPLUGIN_H
#define ATRIAPLUGIN_H
#include ‘‘Plugin.h’’
#include ‘‘PluginProxy.h’’
//Other necessary includes…
class ATriaPlugin: public Plugin
{
public:
//These are required
void input(std:: string file);
void run();
void output(std:: string file);
//Other member procedures…
private:
float* OrigGraph;
std:: string* bacteria;
//Other member variables…
};
__global__ void _GPU_Floyd(int k, float *G, int N);
__global__ void _GPU_Pay(float* D, float* P, int N);
//Other GPU kernels…
#endif
Our source file contains the full implementation of each member function as well as the CUDA kernels, although once again we leave out implementation details. We call both CUDA kernels from run(). Assuming this code resides in the PluMA source tree or within the PLUMA_PLUGIN_PATH PluMA will automatically compile it into a shared object, detecting the compiler based on file extension. Interfacing to PluMA is performed through a plugin proxy (Gamma et al., 1994), which is registered with the PluMA back end through a singleton plugin manager, which accepts our unique plugin name.
#include ‘‘PluginManager.h’’
#include ‘‘ATriaPlugin.h’’
//Other necessary includes…
void ATriaPlugin:: input(std:: string file)
{/*… Read file, initialize variables… */}
void ATriaPlugin:: run() {
//…
//At some point call first kernel
_GPU_Floyd⋘x, y⋙(k, dG, N);
//…
//At some point call second kernel
_GPU_Pay⋘x, y⋙(dG, dPay, (N/2));
//…
}
void ATriaPlugin:: output(std:: string file)
{/*… Final operations, write file… */}
//Other member procedure definitions…
__global__ void _GPU_Pay(float* D, float* P, int N)
{/*… GPU code… */}
__global__ void _GPU_Floyd(int k, float *G, int N)
{/*… GPU code… */}
//Other kernel procedure definitions…
//Proxy
PluginProxy < ATriaPlugin> ATriaPluginProxy
= PluginProxy < ATriaPlugin>(‘‘ATria’’,
PluginManager:: getInstance());
The plugin proxy demonstrates an additional advantage to plugins over scripts, in that they are all executed within one execution time environment, as opposed to a sequence of multiple environments. PluMA plugins additionally can (independent of their language) access a global log file output by a single PluMA execution through the PluginManager, instruct the PluginManager to check for dependencies, and in the future when we implement parallel plugins, send messages to the PluginManager. Note that none of this computational core communication is required to construct a plugin, but it can be useful and easy if necessary.
4 Discussion
4.1 Pipelines and plugins
Through our example P-M16S pipeline for 16S next generation sequencing data analysis, we have demonstrated PluMA’s ability to execute plugins that are heterogeneous with respect to programming language, how to wrap existing software packages as plugins, how to create new plugins, and how to easily use plugins to create flexible and lightweight pipelines that can be run on standard operating systems. As mentioned PluMA’s executable size is only 182K and for P-M16S the average plugin size on disk was 118K, requiring a total of only about 1.1 MB on disk. While runtime memory utilization will inevitably depend on data size to a degree, the design of PluMA ensures that with respect to code only PluMA and the currently executing plugin are required to be in RAM. As plugin dynamic loading and unloading needs to be done only once between these stages, we expect this penalty to be dominated by a plugin’s internal execution time for any reasonably involved algorithm or dataset.
PluMA’s ability to efficiently assemble pipelines where stages can be easily plugged in and out will be important for progress in -omics analysis. If for example in the previous section we wanted to try a different centrality algorithm or even develop our own, we could use any supported language and seamlessly replace ATria with our new plugin in P-M16S with one configuration file change. Once fully tested, that plugin could then join the PluMA plugin pool, and other PluMA users could seamlessly integrate it into their pipelines without modifying their components. Components of these pipelines now become compatible with each other as PluMA plugins, and can be efficiently interchanged without reinventing the wheel which would not likely have been the case with independently developed scripts. Furthermore any of the multiple file conversion plugins in the plugin pool can be integrated as a pipeline stage just as easily and with no loss of generality, as we saw with CountTableProcessing and CSV2GML. PluMA does not even require a user to have access to all supported languages—i.e. a user may not have an NVIDIA graphics card and thus be unable to use CUDA, but PluMA would not require CUDA as long as they do not use CUDA plugins. This is similarly true for R and Perl. We only require the C runtime environment and Python to compile PluMA. When constructing P-M16S we only added plugins in R and CUDA but used Python plugins, implying that we could integrate those plugins into a PluMA pipeline with no knowledge of Python itself, and the analogous case holds for all supported languages. Thus ultimately, P-M16S demonstrates achievement of our goal of providing a flexible, lightweight environment for algorithm developers to construct, test and debug their algorithms with minimal integration overhead. By allowing developers to devote much greater effort to their algorithm, PluMA can help to increase the rate of advancement of microbiome analysis algorithms.
4.2 P-M16S results
As mentioned earlier, the P-M16S pipeline was used to analyze the data from (Kozich et al., 2013). Through our final Cytoscape plugin, produced by PluGen, we were able to visualize both bacterial co-occurrence networks (one for Early and one for Late), as shown in Figure 4. The output GML file from our CSV2GML plugin served as the input network, and we use the output NOA file from ATria as an input table, allowing Cytoscape to color nodes based on their centrality values. Edge thickness indicates weight magnitude (thicker = higher). We ran the Fruchterman-Reingold algorithm (Fruchterman and Reingold, 1991) within Cytoscape to more clearly visualize clusters of bacterial taxa that tend to occur together, and used the abundance of an OTU to determine node size (larger = higher). In both networks we circle and label the five most central OTUs. We also put boxes around highly-abundant OTUs from the Porphyromonadaceae family, which as we recall was found in (Kozich et al., 2013) as a key differentiating OTU between the two datasets.
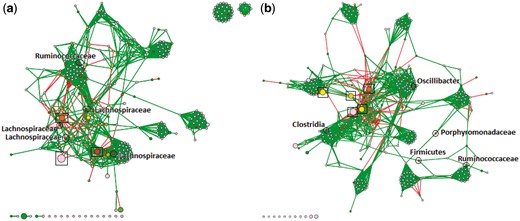
Bacterial co-occurrence networks, built and visualized with our PluMA pipeline, using mouse gut microbiome data (a) before and (b) after obesity. Nodes with the highest centrality have been circled and labeled. Boxes indicate highly-abundant OTUs from the Porphyromonadaceae family. Preliminary results show more abundant Porphyromonadaceae OTUs in the post-obesity samples that negatively correlate with many other OTUs, and more significant roles played by Ruminococcaceae and Lachnospiraceae families in the early samples (Color version of this figure is available at Bioinformatics online.)
We immediately note the differences in the amount of highly-abundant Porphyromonadaceae OTUs between the Early and Late networks, which makes sense given the results of (Kozich et al., 2013). Those five boxed OTUs are the five most abundant OTUs in the Late network, but only three of the five most abundant in Early. All of them appear to be negatively correlated to several other OTUs in the network. Furthermore in the Late network these relations seem to be much more extreme. The Fruchterman-Reingold algorithm seems to ‘push’ these five nodes towards the center, either indicating higher magnitude negative connections or more of them (both appear to be the case). While Porphyromonadaceae is generally considered a ‘good gut bacteria’ for its protective roles (Ferreira et al., 2011), it has also been shown to decrease in human patients that pursue effective weight loss treatments (Clarke et al., 2013). These networks seem to support the theory that Porphyromonadaceae populations tend to be more ‘out of control’ in obese microbiome samples, and that careful control of the population could be a way to combat obesity. Networks like these could also help to estimate potential side effects of treatments that attack Porphyromonadaceae.
The central bacteria also show some interesting results in both networks. Of the top five nodes in the Early network, four are from the Lachnospiraceae family and one is from Ruminococcaceae (this one was the most central). Both of these microbial families have been associated with healthy guts, particularly because of their roles in degrading complex plant material (Biddle et al., 2013). These families did not seem to play as significant a role in the Late network community. Ruminococcaceae was only ranked fourth and Lachnospiraceae did not show up at all, although there were Firmicutes and Clostridia OTUs that could not be classified further. In their places another Porphyromonadaceae OTU emerged as central, and also another from the Oscillibacter genus. Particularly because of its more specific classification level, this latter OTU becomes interesting, and indeed Oscillibacter was featured as a key OTU in differentiating obese samples in another study involving mice (Lam et al., 2012), and also one involving Korean adolescents (Hu et al., 2015).
We summarize these results in Table 3. While these are preliminary theories that require further testing and experimentation, our P-M16S pipeline demonstrates how pipelines that combine ideas in new and creative ways can yield new knowledge and theories to explore, and the reusability of PluMA plugins helps to facilitate this process.
Summary of the importance differences in central nodes between the Early and Late networks in Figure 4
| Rank | Early | Late |
|---|---|---|
| 1 | Ruminococcaceae (Family) | Firmicutes (Phylum) |
| 2 | Lachnospiraceae (Family) | Clostridia (Class) |
| 3 | Lachnospiraceae (Family) | Poryphyromonadaceae (Family) |
| 4 | Lachnospiraceae (Family) | Ruminococcaceae (Family) |
| 5 | Lachnospiraceae (Family) | Oscillibacter (Genus) |
| Rank | Early | Late |
|---|---|---|
| 1 | Ruminococcaceae (Family) | Firmicutes (Phylum) |
| 2 | Lachnospiraceae (Family) | Clostridia (Class) |
| 3 | Lachnospiraceae (Family) | Poryphyromonadaceae (Family) |
| 4 | Lachnospiraceae (Family) | Ruminococcaceae (Family) |
| 5 | Lachnospiraceae (Family) | Oscillibacter (Genus) |
Note: Most notable (in bold) were the general high importance of Lachnospiraceae OTUs in the Early network, and the presence of Oscillibacter and highly-abundant Porphyromonadaceae in the Late.
Summary of the importance differences in central nodes between the Early and Late networks in Figure 4
| Rank | Early | Late |
|---|---|---|
| 1 | Ruminococcaceae (Family) | Firmicutes (Phylum) |
| 2 | Lachnospiraceae (Family) | Clostridia (Class) |
| 3 | Lachnospiraceae (Family) | Poryphyromonadaceae (Family) |
| 4 | Lachnospiraceae (Family) | Ruminococcaceae (Family) |
| 5 | Lachnospiraceae (Family) | Oscillibacter (Genus) |
| Rank | Early | Late |
|---|---|---|
| 1 | Ruminococcaceae (Family) | Firmicutes (Phylum) |
| 2 | Lachnospiraceae (Family) | Clostridia (Class) |
| 3 | Lachnospiraceae (Family) | Poryphyromonadaceae (Family) |
| 4 | Lachnospiraceae (Family) | Ruminococcaceae (Family) |
| 5 | Lachnospiraceae (Family) | Oscillibacter (Genus) |
Note: Most notable (in bold) were the general high importance of Lachnospiraceae OTUs in the Early network, and the presence of Oscillibacter and highly-abundant Porphyromonadaceae in the Late.
4.3 Future work
Several improvements are possible for PluMA. First, we would like to expand its plugin pool to include tools such as newer correlation algorithms [e.g. SparCC (Friedman and Alm, 2012)] and search engines for different databases [i.e. Pathway Tools (Karp et al., 2002)]. Also, we would like to enhance its flexibility by allowing for parameterized specifications in the configuration file, and improve its usability through a Graphical User Interface. PluMA plugins can currently call some computational core functionality and also use features of other plugins in the same language, and we plan to explore SWIG (Beazley, 1996) as a tool for facilitating this compatibility across plugins written in different languages. We could also allow parallelism between independent pipeline stages to improve scalability for larger datasets. Finally, we are currently incorporating Conda package management into PluMA, allowing automatic installation of any PluMA-supported languages that the user wishes to run, to uphold version compatibility over the long haul.
Acknowledgements
The authors acknowledge the help of Astrid Manuel, Arpit Mehta, Daniel Ruiz Perez, Vanessa Aguiar-Pulido, Victoria Suarez-Ulloa, Camilo Valdes and Joaquin Zabalo in trying out the beta version of the pipeline and in many useful discussions.
Funding
This work was supported by the Department of Defense [DoD W911NF-16-1-0494 to GN]; National Institute of Health [NIH 1R15AI128714-01 to GN]; National Institute of Justice [NIJ 2017-NE-BX-0001 to GN]; Florida Department of Health [FDOH 09KW-10 to GN]; Alpha-One Foundation [to GN], Army Research Office Department of Defense [Equipment to GN]; Florida International University [to GN and TC]; Eckerd College [to TC]; and NVIDIA [Equipment to TC].
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}