





Abstract
The tyrosine kinase 2 protein (Tyk2), encoded by the TYK2 gene, has a crucial role in signal transduction and the pathogenesis of many diseases. A single nucleotide polymorphism of the TYK2 gene, SNP rs34536443, is of major importance, since it has been shown to confer protection against various, mainly, autoimmune diseases. This polymorphism results in a Pro to Ala change at amino acid position 1104 of the encoded Tyk2 protein that affects its enzymatic activity. However, the details of the underlined mechanism are unknown. To address this issue, in this study, we used molecular dynamics simulations on the kinase domains of both wild type and variant Tyk2 protein.
Our MD results provided information, at atomic level, on the consequences of the Pro1104 to Ala substitution on the structure and dynamics of the kinase domain of Tyk2 and suggested reduced enzymatic activity of the resulting protein variant due to stabilization of inactive conformations, thus adding to knowledge towards the elucidation of the protection mechanism against autoimmune diseases associated with this point mutation.
1 Introduction
The tyrosine kinase 2 (Tyk2) protein encoded by the TYK2 gene, is a member of the Janus kinases (JAK) family with a crucial role for signal transduction in response to various cytokines, including type I interferon (IFN) as well as proinflammatory and anti-inflammatory cytokines (Langrish et al., 2004; Murray, 2007; Shimoda et al., 2000; Velazquez et al., 1992). Tyk2 is part of the STAT signaling pathway and binds to the type I interferon-a receptor (IFNAR) on the cell surface of IFN-producing cells, thus playing a major role in the pathogenesis of various autoimmune and inflammatory diseases (Kyogoku and Tsuchiya, 2007).
Each of the four JAK proteins consists of a large amino-terminal region, a pseudo-kinase domain and a catalytic domain (or kinase domain). The phosphorylation of the kinase domain (KD) is the activation switch of these kinases (Yamaoka et al., 2004). Various crystal structures of Tyk2 domains have been determined [e.g. crystal structure of an ADP-bound form of its KD; PDB entry code: 4GVJ (Liang et al., 2013), Fig. 1]. The 3D-structure of the KD segment of Tyk2 is divided in two structurally distinct regions, characteristic of protein kinases: the N-terminal lobe (residues: 889–979) and the C-terminal lobe (residues: 985–1176) (Fig. 1). The active site of the protein is located at the crevice created between the two lobes.
![3D-structure of the kinase domain of Tyk2. Ribbon model representation, rendered using coordinates from a known crystal structure [PDB entry code: 4GVJ (Liang et al., 2013)]. β-Strands and α-helices are shown as arrows and ribbons, respectively. Various structural elements and regions corresponding to characteristic features of JAK kinases are shown in color and indicated. Proline 1104 and important for activation tyrosine residues discussed in the text are labeled and shown as stick models](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bioinformatics/34/17/10.1093_bioinformatics_bty556/3/m_bioinformatics_34_17_i781_f1.jpeg?Expires=1716379576&Signature=Jip-LdTT9LI8oZGuk7F2Srlcv5IYVkjIBH3V6isCZ3esE1I7CKEvBKcDVGuFihUbT1NcD1Te3iBKwh6hQi7IEq4hNhHBfwsHBZVj4BJUnyuMU6sSqaLPuHsAM6948fkCsC3JkxHs3lD7~-TCUrLJW1HL24Re4EsVy1LdD1Pug8j~F8tZ9WzIEMkxO78t2hWEylRNGOSmHXCIzUaS9R2kCNf~nxNvplO3jCsDCdj0n3Gap9~l-nwC2d6JwmdDV4WDTV74s4Il92VdeY~pXT-q8eXqGE1ZT6sc~l6viCZaWeKXwrF56bo4kFQ6~DkAxJ-Sv2~tcjGj~LzK8ITj0Ubl3w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
3D-structure of the kinase domain of Tyk2. Ribbon model representation, rendered using coordinates from a known crystal structure [PDB entry code: 4GVJ (Liang et al., 2013)]. β-Strands and α-helices are shown as arrows and ribbons, respectively. Various structural elements and regions corresponding to characteristic features of JAK kinases are shown in color and indicated. Proline 1104 and important for activation tyrosine residues discussed in the text are labeled and shown as stick models
The activation segment of Tyk2 (A-loop), located at the beginning of its C-terminal lobe, contains two sequential tyrosine residues (Y1054, Y1055; Fig. 1), phosphorylation of which is necessary for the activation of this protein (Gauzzi et al., 1996) and subsequent activation of the signaling pathway (Lucet et al., 2006).
Tyk2 is encoded by the TYK2 gene located on chromosome 19p13.2 (Lindqvist et al., 2000). Polymorphisms of the TYK2 gene have been recently found to be associated with various autoimmune diseases as well as endometriosis (Peluso et al., 2013). One of these polymorphisms, namely the single nucleotide polymorphism (SNP), rs34536443 is of particular importance since it has been shown to confer protection against several autoimmune diseases, such as: Multiple Sclerosis (Dyment et al., 2012), Psoriatic Arthritis (Myrthianou et al., 2017), Rheumatoid Arthritis, Systemic Lupus Erythematosus and Inflammatory Bowel diseases (Diogo et al., 2015), as well as protection against endometriosis (Peluso et al., 2013). This polymorphism results in a Pro1104 (Fig. 1) to Ala substitution in the amino acid sequence of the Tyk2 protein and has been shown to affect the enzymatic activity of this protein and therefore the associated cytokine pathways (Couturier et al., 2011; Tomasson et al., 2008). However the atomic details of the underlined mechanism of this, remain unknown.
In this study, we used molecular dynamics simulations, to address this issue.
2 Methods
The molecular dynamics simulations (MD) were performed on the KD domains of both, variant (P1104A hereafter) and wild type Tyk2 (wtTyk2 hereafter) using the GROMACS software package (Hess et al., 2008). The known crystal structure of the ADP-bound form of wtTyk2 [from the Protein Data Bank entry: 4GVJ (Liang et al., 2013); only protein atoms were extracted] and a 3D-model we constructed by in-silico mutagenesis using the VMD program (Humphrey et al., 1996), were used as initial conformations of wtTyk2 and the P1104A variant, respectively. The MD simulations were carried out in explicit water using the gromos96 53a6 force field (Oostenbrink et al., 2004). Periodic dodecahedron boxes of SPC water molecules (Berendsen et al., 1987) extending 10 Å from protein atoms were used to solvate the protein systems, which were subsequently neutralized with counter-ions and optimized by steepest descent energy minimization. Energy optimization was followed by restrained MD simulations where the protein atoms were harmonically restrained to their initial position to allow the solvent to equilibrate. The equilibration step (for a total of 200 ps) was performed for 100 ps under the NVT conditions followed by 100 ps in the NPT ensemble. The temperature and pressure of the systems were kept constant, at 300 K and 1 bar, respectively. The optimization phase was followed by unrestrained MD simulations (MD production runs) in the NPT ensemble. A cut-off radius of 10 Å was employed for all non-bonded interactions in all MD simulations. To overcome sampling problems, multiple (three for each), independent, 100 ns-long MD simulations were carried out for both wtTyk2 and P1104A. The replicas were produced using different, random sets of starting velocities for the atoms. A similar procedure we used in (Voukkalis et al., 2016).
MD analyses were based on various metrics and were performed on the respective MDs of each protein under study, both separately and on average within their sets of MD trajectories.
Convergence of the MD trajectories was assessed by monitoring the root-mean-square deviation (RMSD) of all the backbone atoms from the initial structure, along the MD trajectories.
Cross-correlation maps allow identification of residues moving collectively either in the same or in opposite directions (correlation coefficient values close to +1, or −1, respectively) along MD trajectories. In the present study, cross-correlation coefficients were calculated for the motions of all the Cα atoms of both wtTyk2 and P1104A along their respective concatenated MD trajectories (see above). The 120-ns single trajectories were also used for the corresponding backbone B-factor estimations and structure averaging. RMSF calculations, structure averaging, B-factor estimations and PCA analyses were carried out using related analysis tools of GROMACS, whereas cross-correlation analysis was performed using a related GROMACS tool modified accordingly, in house (C. Zarkadas and M. Vlassi, IB-A, NCSR ‘Demokritos’, unpublished).
Secondary structure analyses made use of the DSSP algorithm (Kabsch and Sander, 1983) and were performed along all the 100-ns MD trajectories. Final models of both wtTyk2 and P1104 (three for each) were obtained by cluster analysis of the last 40 ns of their corresponding MD trajectories. Cluster analysis used the g_cluster module of GROMACS and a backbone RMSD cut-off of 1 Å for two structures to be considered neighbors. The representative structure (structure with the smallest average RMSD from all other structures of a cluster) of each MD simulation was subsequently optimized by the steepest descent energy minimization with flexible water, to obtain the respective final MD models.
PyMOL (The PyMOL Molecular Graphics System, Version 1.7.0.0. Schrödinger, LLC) was used for visualization of trajectories and rendering of molecular model illustrations.
3 Results and discussion
Kinases are highly dynamic proteins and the structural flexibility and concerted motions in their kinase domains, are crucial for the allosteric regulation of the enzymatic activity of these proteins (Huse and Kuriyan, 2002; Kornev et al., 2006; Kornev and Taylor, 2015).
To investigate the dynamics of the Tyk2 structure and the effect of the Pro1104 to Ala change on the structure and dynamics of this protein, we used MD simulations on the KDs of the wild type (wtTyk2) and the P1104A variant of Tyk2, respectively. Because of the stochastic nature of protein MD simulations and to reduce sampling problems, three independent MD simulations for each protein were carried out, and analyzed, as described in Section 2. The simulated systems had reached equilibrium and remained stable, at least, during the last 40 ns of the 100-ns trajectories in all cases and this simulation time range was subsequently used for the analyses of the MD trajectories.
3.1 Fluctuation analysis showed that the structure of the P1104A variant is overall less dynamic compared with wtTyk2
The flexibility of the structure of the two proteins under study was first assessed by means of RMSF analyses (see Section 2). The average backbone RMSF values of wtTyk2 and P1104A residues obtained from their corresponding sets of MD simulations, as well as the RMSF differences of P1104A relative to wtTyk2 along the sequence, are shown in Figure 2A and B, respectively.
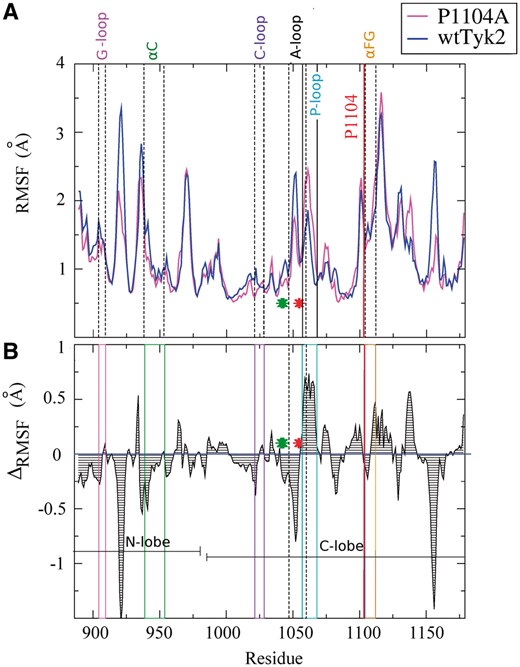
Fluctuation analysis. A). Backbone RMSFs averaged within the respective sets of the three 100-ns MD trajectories of wtTyk2 and P1104A (see Section 2). B). Differences of the RMSF values shown in A, calculated from: ΔRMSF = RMSF (P1104A) – RMSF (wtTyk2). Regions of the amino acid sequence corresponding to characteristic features of protein kinase domains, discussed in the text, are boxed and labeled (refer to Fig. 1, for comparison). Green and red asterisks denote the DFG motif, located just before the activation segment, and the Y1054–Y1055 regions, respectively
As shown in Figure 2, the P1104A variant, and especially its N-lobe and A-loop (bearing the Y1054-Y1055 site), is much less flexible compared with wtTyk2 (negative ΔRMSF values, in Fig. 2B). These observations are better illustrated using coloring of the average MD structures by B-factor values of their backbone atoms obtained from the respective sets of MD simulations of the two proteins (see Section 2), shown in Figure 3. As indicated by low backbone B-factor values (in blue in Fig. 3), the P1104A variant was overall more rigid than wtTyk2.
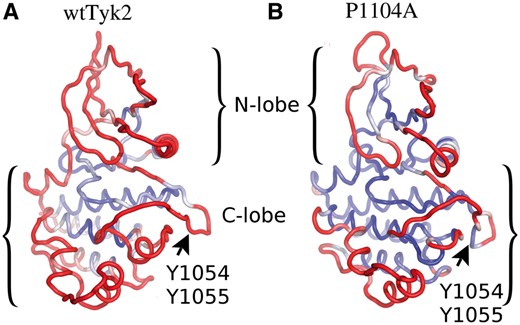
B-factor-based color illustrations of ribbon models of the average MD structures of wtTyk2 (A) and P1104A (B), obtained from their corresponding concatenated MD trajectories. The color scaling according to backbone B-factor values (see Section 2) is from blue to red, for small (=low mean fluctuations; ≤0.6 Å) to large B-factor values (=high mean fluctuations; ≥1.3 Å), respectively. The orientation of the models is as in Figure 1, for comparisons
On the contrary, the N-lobe and a large part of the C-lobe (including the A-loop), appeared to be much more dynamic in wtTyk2, as indicated by higher backbone RMSF (Fig. 2) and B-factor values of these regions (compare B-factor-based coloring in Fig. 3). Interestingly enough, the αFG helix, bearing Pro1104/Ala1104 exhibited equally high fluctuations in both proteins (Figs 2 and 3). Taken together, these observations suggest that the Pro1104 to Ala substitution has long-range effects.
3.2 Cross-correlation analyses revealed a large disruption of correlated motions in the case of the P1104A protein
As already mentioned, concerted movements of various regions of kinase domains are necessary for the allosteric regulation of the activity of protein kinases (Kornev and Taylor, 2015). To address this issue, we first used PCA analysis (see Section 2). Subsequent inspection of the animations of filtered configurations projected on the first eigenvectors (dominant modes of system’s motions) produced by PCA analysis, revealed that the backbone atoms of wtTyk2 moved in a highly concerted manner along the corresponding MD trajectories, as opposed to the P1104A case.
To investigate this further, we calculated cross-correlation coefficients of the motions of the Cα atoms along the corresponding concatenated trajectories for both proteins (see Section 2). Correlation coefficients close to +1 or -1 indicate highly correlated motions (movements of atoms in the same and opposite directions, respectively), whereas correlation coefficients of 0 indicate uncorrelated movements. As shown in Figure 4, the motions of the Cα atoms of wtTyk2 were indeed highly correlated (both positively and negatively) along its concatenated MD trajectories (Fig. 4A and B, top panel). Interestingly enough, these networks of highly correlated Cα motions appeared to be largely disrupted in the case of the P1104A variant (Fig. 4, bottom panel), indicating that the P1104 to Ala replacement affects the communication between various parts of the KD of Tyk2. This, in turn, is expected to affect the catalytic status and the allosteric regulation of the activity of this protein.
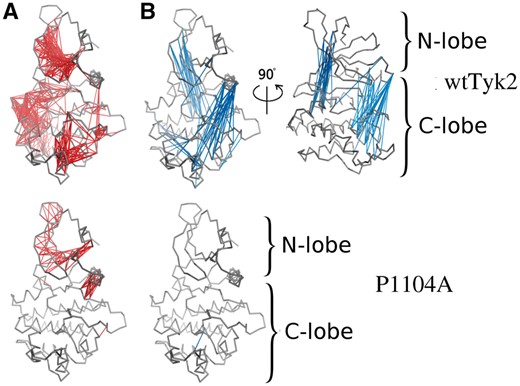
Cross-correlation analysis of the motions of the Cα atoms over the course of the corresponding concatenated MD trajectories of wtTyk2 (upper panel) and P1104A (lower panel). Highly correlated residues (−0.7 ≤ correlation coefficients ≥ 0.7) (identified as described in Section 2) are interconnected by lines colored in red and blue, for positive and negative values of the correlation coefficients of the motions of their Cα atoms, respectively
3.3 Analyses of the MD results showed that the P1104A variant is stabilized in inactive and inadequate for phosphorylation of the activation segment, conformations
To investigate this hypothesis we focused our analyses on various structural features, characteristic of the activation status of protein kinases.
3.3.1 Relative movement of the N- and C-lobes
As indicated by high negative correlation coefficients between the movements of their Cα atoms, the N- and C-lobes of wtTyk2 moved in a concerted manner in opposite directions throughout its respective MD trajectories, whereas P1104A lacked such correlated motions (Fig. 4). Such movements are indicative of a switching between ‘open’ and ‘closed’ conformations of the active site cleft in the case of wtTyk2. This is better illustrated by the distribution of the angle describing the active site cleft, along the individual MD trajectories (Fig. 5A, left panel). As shown in Figure 5A (right panel), the P1104A variant appeared to adopt overall more ‘closed’ conformations, with angle values similar to that of the ADP-bound form of the protein (4GVJ PDB entry) used as initial conformation, in all three corresponding MD simulations. The wtTyk2, on the other hand, adopted more open conformations (rotations of two lobes by more than 10° relative to the starting conformation) with broader distributions of the corresponding angle (Fig. 5A, right panel). Such conformational mobility between the N- and C-lobes is essential for the catalytic activity of protein kinases (Kornev et al., 2006), and its restriction (by the presence of the pseudokinase domain) has been proposed to block the activation of Tyk2 (Lupardus et al., 2014).
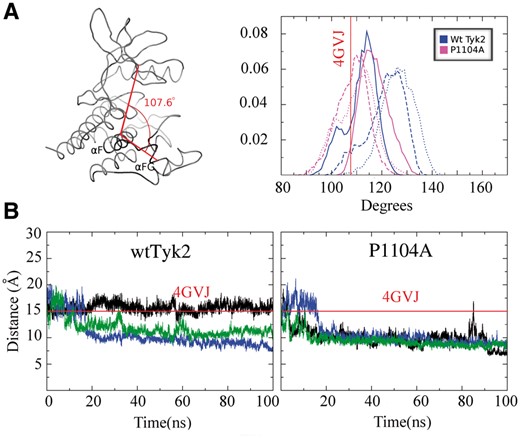
(A) Distribution of the angle between the N- and C-lobes along the equivalent MD trajectories (three for each) for wtTyk2 and P1104A. The definition of the angle is shown on the left panel. The helix αF was chosen as pivot point because of its role as scaffold for the assembly of active protein kinases (Kornev et al., 2008). (B) Monitoring of the distance between the G-loop and the catalytic loop (C-loop in Fig. 1) along the three 100-ns MD trajectories (in black, blue and green respectively) of wtTyk2 (left panel) and P1104A (right panel). Red lines indicate the respective N-C angle and distance in the case of the 4GVJ crystal structure
3.3.2 Distance between the G-loop and the catalytic loop
The distance between the G-loop and the catalytic-loop (C-loop) is another measure of the conformation of the active site of kinases. For example in the case of activated PKA kinase, MD simulations showed that this distance varied up to ∼18.3 Å (open) in the case of the apo-form but was reduced to ∼15.5 Å (closed) in its ATP-bound form (Li et al., 2014). As shown in Figure 5B, this distance varied among the three MD simulations of wtTyk2 (Fig. 5B, left panel), but was reduced and remained constant at much smaller values (<10 Å) compared to 4GVJ, in all three corresponding MD simulations of the P1104A variant (Fig. 5B, right panel), indicating that the G- and C-loops remain constantly in close contact, in the case of the variant. The later observation indicates a persistent placement of the G-loop within the ATP-binding pocket, in P1104A, most probably blocking access of the ATP molecule and therefore hindering the catalytic activity of this variant. In line with this hypothesis, is the observation that the G-, C-loop distance has a similar value (∼9 Å) in the case of a crystal structure of wtTyk2 in complex with an ATP-competing inhibitor-compound bound into its ATP-binding pocket [PDB entry: 3NZ0 (Tsui et al., 2011)].
3.3.3 The ‘molecular break’
The notion of a ‘molecular break’ in their hinge region, regulating the activity of receptor tyrosine kinases, has been introduced by Chen et al. (2007). According to this idea, the formation of hydrogen-bonds between side-chain atoms of three residues on the back of the kinase domain restricts the, required for catalysis, relative motions between the N- and C-lobes of these kinases and is indicative of inactive conformations. The equivalent residues in Tyk2 are: K961, E979 and K1038 located at the N-lobe, hinge region and C-lobe, respectively. Hydrogen-bonds between side-chain atoms of these residues, reminiscent of the molecular break, were observed in two out of the three MD models of P1104A (Fig. 6, lower panel), whereas the situation was reversed in the case of wtTyk2 (Fig. 6, upper panel).
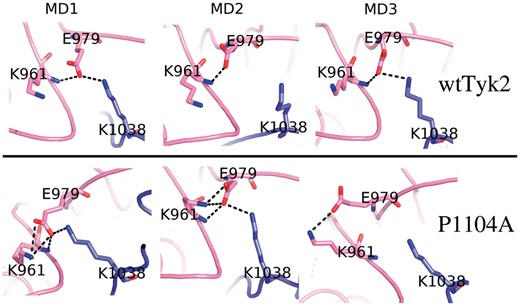
The ‘molecular break’ region. Final models obtained from the corresponding MD simulations of wtTyk2 (upper panel) and P1104A (lower panel), focused on the region corresponding to the ‘molecular break’ (see text). The corresponding Tyk2 residues are labeled and shown as stick models. Hydrogen-bonds are shown with broken lines
Taken together our observations so far suggest that, although the unphosphorylated wtTyk2, studied here, is able to explore various conformations, replacement of P1104 by Ala stabilizes mainly inactive conformations of this protein that block regulation of its activation.
3.3.4 The Y1054-Y1055 region
As already mentioned, activation of Tyk2 requires phosphorylation of tyrosines Y1054 and Y1055 (Gauzzi et al., 1996), which are located at the end of its A-loop (amino acids: 1047–1060) and more precisely, at the beginning of a β-strand (β11), which is part of a β-sheet (β11–β12 sheet) in the 4GJV crystal structure (Liang et al., 2013) (Fig. 1). As shown above (Figs 2 and 3), the segment of the A-loop at the site of Y1054–Y1055 was much less dynamic during the MD trajectories of the P1104A variant compared to wtTyk2 (compare RMSF differences and B-factor-based coloring of the Y1054-Y1055 region in Figs 2 and 3, respectively). To investigate these observations further, we performed secondary structure analyses. Monitoring of the secondary structure along the corresponding MD trajectories is shown in Figure 7.
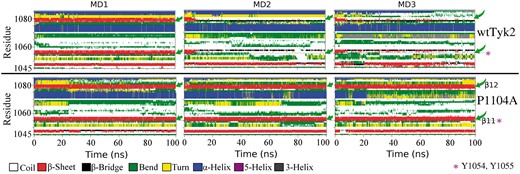
Monitoring of the secondary structure along the equivalent sets of 100-ns MD trajectories of wtTyk2 (upper panel) and P1104A (lower panel). A color definition of secondary structure elements is shown at the bottom of the figure Green arrows point to the position of the initial β11–β12 sheet (see text). The Y1054–Y1055 region is indicated by asterisks, in magenta
As shown in Figure 7 (upper panel) the initial β11–β12 sheet structure (in red) was disrupted and an order-to-disorder (β-sheet to coil; red to white color, respectively) transition occurred in this region after the first 10 ns in two (MD2, MD3) out of three MD simulations of wtTyk2 (Fig. 7, upper panel). This observation, in conjunction with the importance of structural disorder in protein recognition and phosphorylation (Iakoucheva et al., 2004), is in perfect agreement with the ability of wtTyk2 to get phosphorylated at these tyrosines and therefore activated, as expected.
On the contrary, the β11–β12 sheet was preserved during all three MD simulations, in the case of P1104A (in red Fig. 7, bottom panel). This observation, in conjunction with low fluctuations of the region (Figs 2 and 3), suggests that, in contrast to wtTyk2, the activation segment of P1104A adopts conformations of the phosphosite (Y1054-Y1055) inadequate for phosphorylation, thus preventing activation of this variant. This hypothesis is in perfect agreement with experimental data showing reduced phosphorylation and enzymatic activity of Tyk2 to be associated with the rs34536443 polymorphism (Couturier et al., 2011; Dendrou et al., 2016; Tomasson et al., 2008).
4 Conclusions
In this study we used molecular dynamics simulations to explore the structural consequences of the Pro1104 to Ala substitution, associated with the rs34536443 polymorphism of TYK2 gene, on the resulting Tyk2 protein, and therefore its function.
Our MD results revealed that this single amino acid change has long-range effects that restrict the dynamics of the Tyk2 variant in favor of inactive conformations that most probably switch-off the activation process of this protein, thus blocking the regulation of its enzymatic activity and therefore the associated signal transduction. This hypothesis is in perfect agreement with previous studies linking reduced phosphorylation and enzymatic activity of Tyk2 and attenuated signal transduction with the rs34536443 polymorphism (Couturier et al., 2011; Dendrou et al., 2016; Tomasson et al., 2008).
In total, the results presented here, are in line with the notion that reduced Tyk2 kinase activity may be the mechanism for protection against autoimmune diseases and add to knowledge in support of the idea of targeting Tyk2 and its pathways as a therapeutic approach against these diseases (Couturier et al., 2011).
Funding
We (IB-A; NCSR ‘Demokritos’) acknowledge support of this work by the project ‘Target Identification and Development of Novel Approaches for Health and Environmental Applications’ (MIS 5002514) which is implemented under the Action for the Strategic Development on the Research and Technological Sectors, funded by the Operational Programme ‘Competitiveness, Entrepreneurship and Innovation’ (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}