





Abstract
Over 25% of protein structures possess unresolved fragments. On the other hand, approximately 6% of protein chains have non-trivial topology (and form knots, slipknots, lassos and links). As the topology is fundamental for the proper function of proteins, modeling of topologically correct structures is decisive in various fields, including biophysics, biotechnology and molecular biology. However, none of the currently existing tools take into account the topology of the model and those which could be modified to include topology, demand experience in bioinformatics, protein topology and knot theory.
In this work, we present the GapRepairer—the server that fills the gap in the spectrum of structure modeling methods. Its easy and intuitive interface offers the power of Modeller homology modeling to many non-experts in the field. This server determines the topology of templates and predicted structures. Such information when possible is used by the server to suggest the best model, or it can be used by the user to score models or to design artificially (dis)entangled structures.
GapRepairer server along with tutorials, usage notes, movies and the database of already repaired structures is available at http://gaprepairer.cent.uw.edu.pl.
Supplementary data are available at Bioinformatics online.
1 Introduction
The function of a protein is intimately connected to its structure. However, during last years it has become evident that not only the fold itself but also the topology of the backbone plays a crucial role in biophysical and biological properties of the proteins (Christian, 2016; Dabrowski-Tumanski and Sulkowska, 2017a; Haglund et al., 2017).
In fact, the protein backbone can be tied up into a knot (non-self-intersecting curve in 3D space) or a slipknot (the shoelace-type knot, where one loop is partially threaded through a second twisted loop) (King et al., 2007; Sułkowska, 2012). It can also form a lasso—a structure that contains a covalent loop (closed by e.g. a disulphide bridge), pierced by at least one free end of the structure (Gierut, 2017; Niemyska, 2016), or a link (with two covalent loops piercing each other) (Dabrowski-Tumanski and Sulkowska, 2017b). These structures are schematically presented in Figure 1.

The general idea of GapRepairer. Protein structure with two gap fillings (black and gray, left panel) differing in topology. The one-click approach leads to proper gap filling and determines entanglement type (knot, slipknot, lasso or link) of templates and results (Color version of this figure is available at Bioinformatics online.)
In total, up to 6% of protein chains deposited in PDB possess a non-trivial topology. They can be artificially disentangled upon even a fold-preserving variation in chain arrangement. On the other hand, such chain movement can artificially entangle topologically trivial structures. This, in turn, may affect the free energy landscape of proteins, e.g. the mechanical or thermal stability (Haglund, 2014; Sułkowska et al., 2008) or folding time (King, 2010). Therefore, topologically correct models are crucial for any researcher working in the field of protein structures.
The problem of determining the correct topology is especially evident in the case of proteins with unresolved fragments. Currently, over 25% of protein structures deposited in Protein Data Bank contain one or more gaps, and this number is predicted to increase due to high interest in low-resolution techniques, such as cryo-EM. Moreover, even the existing models (due to low resolution) may already be affected by the wrong topology (either artificially knotted or unknotted). In fact, any dubious fragment of the chain (not only missing part) should be verified for correct topology. The need for topology control spans, therefore, a wide range of problems, from X-ray, NMR study or cryo-EM reconstruction, to de novo modeling, to molecular dynamics simulations or studying mechanical properties of proteins, and drug discovery. However, currently no modeling tool takes the topology into account, nor any method of topology control has been established yet. Some algorithms to find simple knots or pokes (similar to slipknots) have been used to test results in CASP competitions [e.g. Knotfind, Pokefind (Khatib et al., 2006, 2009)]. However, these algorithms have never been used to improve protein reconstruction. Utilization of homology modeling is restricted, due to the necessity of finding a set of topologically correct templates. Therefore, although of huge relevance, topologically valid modeling is accessible only for experts in bioinformatics, protein geometry and knot theory—hence only for a small group of researchers.
GapRepairer is a server designed to bridge this important gap and to deliver the topological assessment of refined structure to every user. It provides easy, intuitive, but comprehensive visualization of structures and entanglement for generated models. The topological assessment is based on the result, that the protein topology is better conserved than the sequence (Sułkowska, 2012). Hence, the target’s topology is predicted to match the topology of the closest homolog. This, in turn, allows selecting only the templates with the expected knot, slipknot and lasso (e.g. when different topological types are represented in one Pfam family). After homologous modeling, the topology of obtained models is assessed based on the known protein entanglement types and the models with unlikely topology (e.g. knots other than of twist type, too complex topology or fingerprint) are pointed out.
The same algorithm may be used to reconstruct fragments of a protein based on the template with the desired topology. The automated batch analysis of files makes GapRepairer useful for CASP or CAPRI competitors. Finally, for more advanced users, GapRepairer gives additional options, such as various template selection methods (like structural homologues), or modeling in the presence of a neighboring chain, which is a novelty among all on-line tools. To generate models the server uses the versatility of Modeller software (Webb and Sali, 2014). As the main aim of the GapRepairer is to conduct the topological analysis (unavailable in any other tool), it does not include other structure validating markers, such as RMSD, GDT. Nevertheless, to additionally assess the models, the user can display the electron density map, if available.
In general, the server does not modify already present structure more than absolutely necessary—thus is not correcting any geometrical errors of the original coordinates. As such users are invited to verify their models with tools designed e.g. the wwPDB validation report (Berman et al., 2003). When such geometrical errors are spotted the server allows to remodel given subchains without taking into account original coordinates. To illustrate its power, the GapRepairer includes also a database of modeled interesting structures from servers which collect entangled proteins—KnotProt (Jamroz et al., 2015), LassoProt (Dabrowski-Tumanski et al., 2016a) and LinkProt (Dabrowski-Tumanski, 2017).
2 Materials and methods
2.1 Topological analysis of a given protein
Knots and slipknots are detected according to the probabilistic algorithm (Millett et al., 2013; Sułkowska, 2013). The entire information about the protein topological complexity is presented as the knot fingerprint matrix. The (i, j) entry of the matrix denotes the topology of the i – j subchain. In general, the topology of the subchain is dependent on the chain closure, hence for each subchain we perform 200 random closures and calculate the statistical probability of each knot obtained. All knots whose probability is larger than 50% are shown on the fingerprint matrix. The type of knot and its probability is presented respectively via different colors and intensities. The lasso structures are detected according to the minimal surface analysis (Niemyska, 2016). The lasso type is assigned based on the number and the directions of piercings through such surface by the protein tail.
2.2 Proteins in the database
The database contains more than 100 reconstructed structures chosen due to their unusual topology or classified as artificially entangled by KnotProt, LassoProt and LinkProt databases. Atom coordinates are taken from PDB deposited.cif files.
2.3 Graphical presentation and technical details
Protein structures, topological nontriviality location along the protein backbone, the detected topology and the minimal surfaces are displayed using JSmol (HTML5/JavaScript). The charts are generated using Plotly. The database is written in Python, dynamically generating HTML pages using Apache2 with WSGI. The data are stored using the SQLite3 database. Information about proteins is downloaded from the PDB using RESTful services. The service is installed on multicore Linux nodes.
2.4 The gaps are filled using Modeller software
The following options are used to model structures: md_level: very_slow; repeat_optimization: 3; max_molpdf: 1e6; max_var_iterations: 500; and for one of the models deviation set to 0 (Webb and Sali, 2014). These parameters were chosen as the best to reconstruct proteins with potentially non-trivial topology, as well as to not introduce artificial crossings into protein structure. Proteins deposited in the GapRepairer database were used as a testing set.
3 Results
GapRepairer consists of two parts—the core is the server (‘Repair my structure’ option) and the list of jobs (‘Jobs’). The second part is the database of already repaired structures (‘Browse database’). The server is accompanied by a complete on-line manual. In the applications section, we discuss a few examples of the most interesting reconstruction of proteins deposited in the database and general applications of the server. More advanced options, e.g. how to rearrange fragments of protein backbone to change its topology, are presented via movies, gaprepairer.cent.uw.edu.pl/cgi-bin/example_knot and gaprepairer.cent.uw.edu.pl/cgi-bin/example_link.
3.1 “Repair my structure” and “Jobs” tabs
To model the gaps and assess the topology, it is enough to enter PDB ID of the structure in the ‘Repair my structure’ tab or upload the coordinates file in the PDB format along with appropriate FASTA sequence (Fig. 2). By default, the gaps and templates are identified automatically. The user can also model the chain of interest in the vicinity of neighboring chains—these are treated as a spatial restraint. In such case, only FASTA sequence for the modeled chain should be supplied. To our knowledge, modeling in the presence of neighboring chains option is unique among all web-servers.
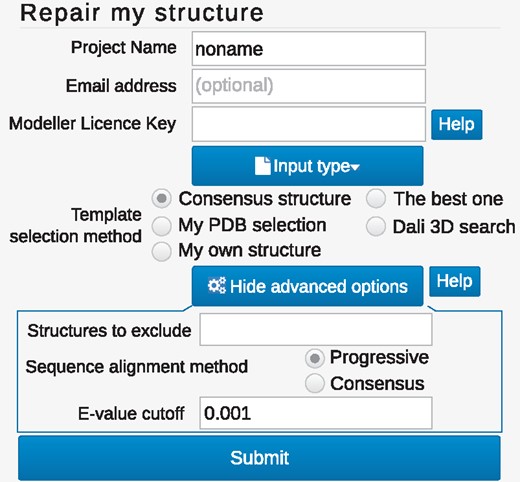
The view of the repair form. The minimal data needed for job submission is the protein structure and sequence which can be specified upon ‘Input type’ button. To facilitate modeling, the user can influence template selection via changing the search method or adjust the method options in ‘Advanced option’ (displayed in the bottom box)
The default option for homologues search (the Consensus option), relying on the list of homologues found by PSI-Blast algorithm, should be sufficient in most cases. However, to enhance the modeling, one can benefit from various template selection approaches (Fig. 2). If only one homologue is enough for structure rebuilding, the user can choose The best one option. For proteins with no clear sequential homologues, viable templates may be hard to distinguish. In such cases, an alignment based on structural similarity found using DALI database (DALI 3D search) can yield better results. Alternatively, the user can upload his own PDB file (My own structure). Each generated set of templates may be downloaded later, therefore GapRepairer may also serve as a tool for (topologically validated) template selection for homology modeling. When the target comes from the RCSB PDB, we provide the user with a link to the wwPDB validation report for the original database submission. Users can name their job, select to hide them in the job queue, and, by supplying their e-mail address, request to be notified upon job finishing. E-mail address is stored until the job finishes, all other data about the jobs are stored for two weeks.
Advanced options. For more advanced users, the GapRepairer server offers other options to influence e.g. the template search. In particular, the user can include or exclude certain structures—this option is useful while assessing the topological correctness. In such case, the user should remove questioned part of the structure and then rebuild it with GapRepairer. To achieve reliable modeling results, the original structure should be excluded from possible templates. Moreover, the user can modify the default e-value cutoff for PSI-Blast search, to increase or decrease the number of potential templates or model missing tails.
All the Modeller input files (which include both cleaned template files and filled-out python script) can be downloaded. This enables to modify the code and to run the modeling on the user’s machine if the Modeller license is not supplied. Finally, the server can be invoked from the command line. This option is designed especially for automated gap filling in a large set of proteins, useful e.g. during CASP/CAPRI competitions for crystallographers, NMR and CryoEM researchers. The on-line documentation includes detailed explanations of form fields and an example of a bash script for batch processing of structures.
3.2 Protein model presentation
One of the aims of GapRepairer is to provide the most user-friendly tool in terms of analyzing modeling results. The results page for every job is divided into three modules. The main part is the Structure presentation with the five best-predicted models superimposed using JSmol (compatible with most browsers—Fig. 3). This allows to use the standard JSmol options (rotate, zoom, etc.). The previously known parts of the chain are colored in gray, and the gap fillings are given in shades of the same color. Any additional chains (if present) are in black.
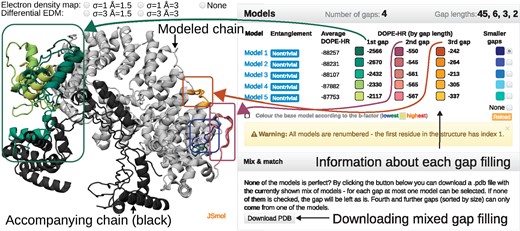
The example of the job results page. Left panel—interactive visualization of the reconstructed structure of a protein with PDB ID 4WZS (chain C): modeled chain (input data) in gray, the shades of the same color denote different gap fillings, accompanying chains in black. Above the visualization, the radio buttons allowing to display electron density map. Right panel—a table with basic information regarding each model, including information about entanglement (the second column) and the DOPE-HR score for the whole structure and for each gap separately. The colors in the table match the colors of gap fillings (as shown by arrows). Lower right is the Mix & match panel allowing the user to create a structure containing gap fillings from multiple proposed models (Color version of this figure is available at Bioinformatics online.)
Details of the models are summarized in the table including information on topological nontriviality and the DOPE-HR potential for the whole structure and each gap separately (the lowest value usually indicates the best model, Fig. 3). The display of each gap filling can be turned on/off by clicking on the appropriately colored square. The color of the square corresponds to the color in the structure presentation. The neighboring chain(s) (black) can be turned off. For improved analysis of the structure—Color the base model according to the B-factor—option is available.
As a novel feature, the user can create a mix of proposed gap fillings (e.g. take the first gap filling from Model 2 and the second gap filling from Model 1). To do so the user has to choose the desired loop filling fragments (by clicking appropriately colored squares in the table) and then Download PDB in the Mix & match panel. To our best knowledge, GapRepairer is the only service that offers such an option.
To validate the models obtained, the user can display the electron density maps (2mFo-DFc and mFo-DFc, both in two versions) or density maps for Cryo-EM structures, if available at EMBL-EBI EDS server. If the target structure provided by the user containes crystallograpic cell and space group, this data is kept in the final model, which allows users to submit them to the wwPDB Validation Service. If available, a link to the original validation report for the RCSB database submission is also given.
Comprehensive topological analysis for each model is presented in Entanglement section. The details about topology are summarized in the table and visualized on the structure (Fig. 4). The table shows the type of entanglement (knot, slipknot or lasso), and its class (the neighboring blue box). When knots or slipknots are detected the location of their knotted core (the smallest knotted piece of chain), and, if applicable, slipknot loop (piece of the chain forming the threaded loop) are given. The total complexity of the model is presented in the form of a knot fingerprint matrix.
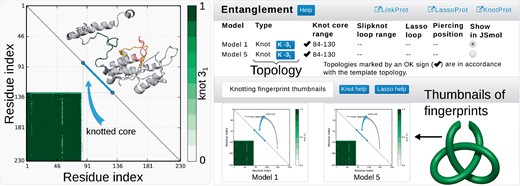
Exemplary, simplified output of the topological analysis. Right panel—the table indicating topological details about models. Columns show the model name, the entanglement type, location of (slip)knot along sequence and lasso data. The structure can be visualized in JSmol. Below, the thumbnails of the knot fingerprint matrices are shown. Left panel—enlarged knot fingerprint showing the knot core, slipknot loop and slipknot tail (as in the table). The probability of obtaining 31 knot is given by the color bar. Inside the matrix, the structure of the protein (above diagonal) along with corresponding knot core (below diagonal) cut out from the structure (Color version of this figure is available at Bioinformatics online.)
The matrix presents the type and probability of knot formation for each subchain. It allows prescribing the total entanglement, which is highly conserved and characteristic for a given function (Sułkowska, 2012) (hence enabling function prediction) or to localize the knotted core or slipknot loop. Such structural elements (e.g. knotted core borders) may reveal functionally important residues (Dabrowski-Tumanski et al., 2016b) or parts differing in effective stiffness (Wang, 2013).
When the lasso topology is detected, its structural details are shown in following columns. This includes the indices of the most complex lasso covalent loop (position of bridging residues) and indices of loop surface piercing residues (with a sign indicating the direction of the piercing). Depending on the number of piercings through this surface, their orientation and the piercing tail, different lasso types are prescribed. Currently, over 30 different lasso types are known according to LassoProt. Moreover, the piercing through the auxiliary surface is also presented using barycentric view (Niemyska, 2016) (Fig. 5). The barycentric view can be especially useful when analyzing self-intersecting surfaces.
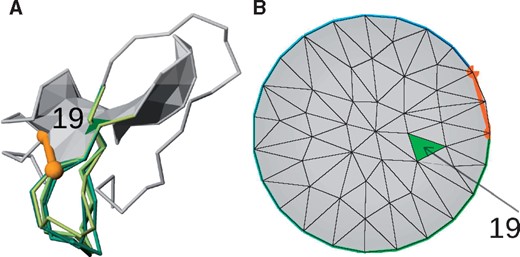
Visualization of lasso topology, in this case, single lasso L1 of exemplary chain (protein with PDB ID 5C67 chain C). (A) The JSmol visualization of the minimal surface spanned on the covalent loop. The loop is closed by the disulphide bond (orange cylinder with beads). The surface is pierced by the chain through the blue triangle. Different gap fillings are presented with different shades of green. (B) The barycentric view of the surface facilitating spatial location of pierced triangles relative to each other and to bordering covalent loop. The pierced triangle is shown in green. The index of piercing residue is 19 (Color version of this figure is available at Bioinformatics online.)
To facilitate understanding, the topology can be visualized in the protein display upon ticking appropriate model in Show in JSmol column. This changes the display in the JSmol presentation to exhibit explicitly the knotted or slipknotted core (for knots or slipknots), or the minimal surface spanned on the covalent loop with colored pierced triangles (for lassos).
Templates, DOPE plot, Download and Log tabs contain additional information about the modeling process and templates used. The interactive DOPE plot given for each model shows on hover the index and the type of residue along with the potential in this location. In the Templates tab, all the templates selected for modeling are presented and aligned. The templates are supplemented by their entanglement type and basic alignment information (sequence identity, total gap coverage, total gap identity, RMSD vs gapped structure). Amino acids in the alignment are colored according to the I-Tasser scheme. The user can also download the models, the alignment or the fingerprint matrices in text format in the Additional Downloads tab. The modeling process can be inspected in the Log tab.
3.3 Protocol
The protocol used follows the procedure: Files cleaning and conversion → structure verification → alignment → template selection based on the topology and gap coverage → modeling → topology verification.
Input files. From user-supplied files, non-protein residues are removed based on a combination of ‘HETATM’ lines, location within the chain, and distance from other residues. RCSB files are cleaned based on non-polymer ligands list available from RESTful services. Sequence overhangs, terminal regions with no structure assigned are kept only when an advanced option ‘Model missing tails’ is specified.
Structure verification. The structure verification is two-step: first, we ensure that sequence provided for each gap is sufficient to fill the spatial distance between the gap flanking residues (we assume at most 5 Å for each residue in the straight line distance). Second, distances between Cα atoms in the existing structure are checked with the same 5 Å criterion.
Alignment. Sequence alignments (including target sequence-to-structure alignments) are performed using a modified Needleman-Wunsch algorithm to ensure proper gap conservation.
Template selection. Templates selection depends on the option chosen by the user. Templates are aligned and sorted based on the sequence similarity in the gap regions to the target. The topology of the best template is used as a reference, and all the templates with a different topology are discarded. Remaining templates are marked as useful for a given gap if they have at least a 30% coverage of the gap, and they cover some residues which have not yet been covered by other templates.
Modeling. Two-residue-long gaps, missing atoms, residues not covered by templates and tails are filled de novo. Longer gaps are modeled based on templates previously marked as necessary. First, there are at most 3 attempts to make a model based on the alignment (AutoModel class, with each gap broadened by two residues). Then, each gap with the local DOPE score above 1000 is remodeled using LoopModel routine (5 tries, the best scoring one is kept). If the resulting model does not pass the Cα distance validation (and previous attempts were template-based) a de novo modeling of all gaps is attempted.
Topology verification. Each of the correct models is subjected to knot- and lasso-detection protocol indicating the structures with most possible topology.
3.4 “Browse database” tab
A database of already repaired structures is stored under Browse database tab. The database is divided into ‘Knotted’ (and slipknotted), ‘Lasso’ and ‘Trivial’ lists. For each entry, the topologies found in all models are listed, with implausible entanglements (according to our experience with entanglement structures) marked in red. These structures need more careful modeling, with a spatial rearrangement of the known amino acids to reconstruct backbone with higher fidelity. Among properly reconstructed proteins, some models are very interesting since we found them uniquely knotted or unknotted. E.g. protein with PDB ID 3OC3 chain A should be knotted according to the best-received model. However, after remodeling one of the flexible loops, the protein chain can be modeled in a different way, leading to an unknot. This implies that these models need further experimental investigation. More examples are discussed in the Applications section.
3.5 On-line documentation
The server is supplemented by the on-line documentation containing the detailed description of the protocol, possible uses, input files preparation, analysis of results, command line access and statistics. Everything is supported by an extensive set of examples. Advanced reconstruction of proteins is supported with movies.
3.6 Applications
The entanglement appears in protein modeling in various ways giving rise to many applications of GapRepairer. The most fundamental is topologically valid gap modeling. The server may also be used to assess the topological correctness of the structure. This is especially important in the case of low-resolution structures (such as CryoEM) and in structure prediction competitions (e.g. CASP, CAPRI). Another application is to generate a topologically valid list of homologues (either structural or sequential). Finally, the user can utilize non-entanglement-based functions of GapRepairer unique to this server, such as the modeling of a chain in the neighborhood of other chains (important for domain-swapped structures), or modeling chains with exceptionally large gaps.
Topologically valid gap modeling. For some structures, the easiest way of modeling is not the best one, as it can artificially (dis)entangle the target. E.g. the original structure of glycohydrolase (PDB ID 3SIJ) possesses a deep 31 knot. However, the non-trivial topology stems from the location of the 9-residue-long gap. Modeling this gap with GapRepairer correctly disentangles the model (Fig. 6A). Similarly, the hydrolase with PDB ID 2D7D, after a proper modeling changes topology from deep 41 knot to unknot. Modeling CryoEM structure of human 26 s proteasome (resolution 6.8 Å, PDB ID 5LN3, chain N) also disentangles the protein. The modeling can also change the chirality of the knot and reduce the complex topological fingerprint, as in the case of human hydrolase with PDB ID 4ZG6. The gapped structure features a left-handed knot, but after gap filling, it turns out that it has the right-handed knot (Fig. 6B).
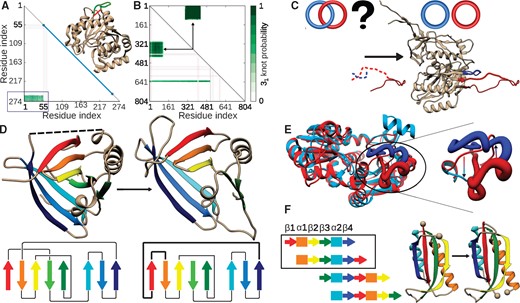
Possible utilization of GapRepairer server. (A) Disentangling of an artificially knotted protein with PDB ID 3SIJ—the straight interval joining gap ends (red stripe in the structure) results in 31 knotted protein, as shown in the matrix fingerprint (highlighted by blue rectangle). The correct way of gap modeling is shown with the green curve on the structure. (B) Change in the fingerprint and topology upon gap modeling for the protein with PDB ID 4ZG6. Before modeling (below diagonal) two knotted regions can be spotted. After gap modeling (above diagonal), only one portion of the chain is 31 knotted. (C) Assessing topological correctness. For the potentially Hopf-linked protein with PDB ID 3J70, removing and remodeling of parts of the loops (dashed lines in left panel), results in unlinked loops (right panel). The topology in each case is shown schematically above the structure. (D) Validating crystallographic data—remodeling parts of the protein with PDB ID 2XKL (left panel) reveals an incorrect connection between β-strands (right panel). For each case, the scheme of β-strands connection is shown below the structure. (E) Search for a topologically valid template—the structures of ATC (red) and OTC (blue) are almost perfect structural homologues, yet they differ in the location of pieces of chain in one part, shown as the thick structure and enlarged in the right panel. Interchanging the parts according to the arrows shown in the right panel changes the topology of the protein. (F) The idea of circular permutation of protein fragments. The right panel shows exemplary structures corresponding to the scheme in the frame
Even though GapRepairer is not optimized to reconstruct membrane proteins, it properly models loops based on structural and topological assumptions when homological chains are unknown. E.g. modeling amino acid transporter (PDB ID 5LLM, chain A, 4 gaps up to 23 amino acids) changes its topology from knot to slipknot characteristic to all of the members of Sodium: dicarboxylate symporter family. Another membrane protein (PDB ID 5L25) has 6 gaps in total, one comprising 94 residues. In this case, careless modeling generates the complex topological fingerprint, with unnatural 82 knot. Proper modeling of this loop results in regular slipknot motif. Furthermore, reconstruction (using only structural homologues) of the membrane protein with PDB ID 4KJS chain A reveals the knot and therefore the first deeply knotted membrane protein family (Jarmolinska, 2017). Figures in Supplementary Material show the essential geometric features of these proteins before and after modeling.
Assessing the topological correctness is crucial for already known structures with dubious fragments, e.g. coming from low-resolution techniques, such as CryoEM. For example, the human immune system related protein (PDB ID 3J70, from CryoEM) contains, according to the model, two linked covalent loops. However, after cutting out a part of each loop and remodeling, the loops turn out to be unlinked, which stays in accordance with all experimentally determined homologues (Fig. 6C). Similarly, the structure of methyltransferase (PDB ID 1OY5) is unknotted, although all proteins from this family form a deeply knotted structure. After the remodeling of this protein, a structure with a deep 31 knot is recreated. Finally, the lasso structure may also disclose improperly crystallized protein. The murine lipid transport protein (PDB ID 2XKL) has a L4 topological type (with four piercings through the surface spanned on the covalent loop, not found yet, according to LassoProt). Our remodeling reveals that the β-sheets in the crystal structure were joined in a wrong order (Fig. 6D). Such topological assessment is decisive in prescribing the function and the properties of a protein, especially as the function of a protein can be guessed knowing the function characteristic for such topological motif (Niemyska, 2016; Sułkowska, 2012).
The GapRepairer can be also used to design a new architecture of the proteins, e.g. to design entangled proteins, or disentangle the topologically non-trivial ones by the addition of a loose loop. This can be a useful technique in understanding the role of knots (King, 2010; Yeates et al., 2007) or lassos (Haglund et al., 2017) (by comparison of different topology homologues). The ability to design topologically non-trivial structures can be as well used to engineer highly stabilized proteins (Ghosh et al., 2015) or to design intradomain linkers (Scalley-Kim et al., 2003).
Template selection is fundamental in any homology modeling or homology-based function prediction. However, even the close homologues can differ in topology as e.g. the pair Acetylornithine TransCarbamylase (knotted) and Ornithine TransCarbamylase (ATC/OTC) (unknotted). Only the GapRepairer, using its topological analysis can distinguish these cases and select relevant close homologues (Fig. 6E).
Other uses. The GapRepairer is also equipped with functions, not necessarily connected with the protein topology. The ability to repair one chain in the presence of the second makes it a unique on-line tool capable of treating domain-swapped proteins. It also allows GapRepairer to model gaps in many-chain systems. The GapRepairer has no restriction on the gap size if only enough homologues are present. This allows one to model a structure with circularly permuted fragments of the protein (Fig. 6F), one of the techniques used to investigate the protein energy landscape (Lindberg, 2006). Finally, because of the DALI database utilization, GapRepairer is the only webserver which can cope with structures with no sequential homologues, if there are some structurally similar chains.
3.7 Comparison with other servers
Currently, there are many available servers allowing online structure repair [ArchPred (Fernandez-Fuentes et al., 2006), ModLoop (Fiser et al., 2000), Rosetta (Rohl et al., 2004)], which however can cover only the easiest, small, one-gap cases. None of the web-tools known to us can model many gaps of arbitrary size, or include the presence of neighboring chains. This can be done only with the use of the most sophisticated tools, such as Modeller software (Webb and Sali, 2014). However, with increasing complexity of the problem, a difficulty level of the usage increases.
The power of GapRepairer lies in its ability to distinguish topology (knots, slipknots and lassos) both of the templates and of the models. This provides a unique possibility to obtain the topologically valid models or the topologically filtered list of homologues. Conversely to most servers, the whole process is fully transparent (including the access to templates, their DOPE potential and alignment) and the results are presented in a clear way enabling direct on-line structure inspecting. The job submission process is greatly simplified, however, GapRepairer has a few exclusive options, such as a search through DALI structural database, possibilities to include or exclude chosen structures, or mixing obtained models.
Furthermore, most of the similar servers use de novo modeling, starting from the sequence only [e.g. I-Tasser (Zhang, 2008), Swiss-Model (Schwede et al., 2003), HHPred (Söding et al., 2005), etc.]. Hence, the details of the resolved parts are not included and computations are much more complex. On the other hand, the servers devoted to loop modeling (e.g. ArchPred) give the user not enough freedom in parameter adjustment.
4 Summary
In this article, we presented the GapRepairer server—the only tool known to us, which determines topology (knots, slipknots, lassos and links) of templates and models. Such knowledge when possible is used by the server to suggest the best model, or it can be used by the user to score models or to design artificially (dis)entangled structures.
The server is easy to use even for inexperienced researchers, while at the same time it contains many easily adjustable options addressed to the more advanced users. The server proposes five models, each with the graphical analysis facilitating the description of entanglement. In particular, the knot fingerprint matrices and barycentric views are given. The extent of the topologically non-trivial fragment of the chain—(slip)knotted core or the surface spanned on the covalent loop—can be explicitly exhibited on the protein structure. Also, any crystallographic data present within the original structure file are preserved—this allows a hassle-free upload to verification tools (like wwPDB service).
Moreover, the server enables to model proteins without restriction on the number of the gaps, in the vicinity of other chains, and based on various sequence and structural template selection methods. The advanced options enable to design artificially entangled or disentangled structures, new folds, circularly permuted fragments of proteins, interdomain linkers or domain-swapped structures.
The GapRepairer posses the database of modeled protein chains, which were classified as artificially entangled or disentangled based on other databases. We present some unique models of proteins, which lead to the identification of new knotted families and postulate a disentangled topology in some other proteins, including lasso and linked proteins. Moreover, after reconstruction, a new protein fold is also suggested based on the topological analysis. Some of these proteins do not have homologous structures, thus they need further experimental validation.
The server possesses the possibility to upload the structure from the command line which makes it especially useful for any automated validation of the protein topology, e.g. during CASP, CAPRI competition or CryoEM methods. We are sure that due to its intuitiveness and versatility the GapRepairer will be an important bioinformatical tool used by a broad spectrum of researchers.
Funding
This work was supported by the National Science Centre [#2012/07/E/NZ1/01900 to JIS, #2016/21/N/NZ1/02848 to PD-T, #2014/15/B/ST6/05082 to MK] and Polish Ministry of Science and Higher Education [#0003/ID3/2016/64 Idea Plus to JIS].
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}