





Abstract
Comparative studies of protein sequences are widely used in evolutionary and comparative genomics studies, but there is a lack of efficient tools to identify conserved regions ab initio within a protein multiple alignment. PROBE provides a fully automatic analysis of protein family conservation, to identify conserved regions, or ‘blocks’, that may correspond to structural/functional domains or motifs. Conserved blocks are identified at two different levels: (i) family level blocks indicate sites that are probably of central importance to the protein’s structure or function, and (ii) sub-family level blocks highlight regions that may signify functional specialization, such as binding partners, etc. All conserved blocks are mapped onto a phylogenetic tree and can also be visualized in the context of the multiple sequence alignment. PROBE thus facilitates in-depth studies of sequence–structure–function-evolution relationships, and opens the way to block-level phylogenetic profiling.
Freely available on the web at http://www.lbgi.fr/∼julie/probe/web.
1 Introduction
Comparative studies of protein sequences are widely used in evolutionary and comparative genomics studies, e.g. to infer structural or functional features or to predict the effects of genetic variants (Giles and Emes, 2017). Traditionally, these comparisons were performed for a single protein family, but recent developments have led to systems-level studies of functional modules, such as macromolecular complexes or signaling pathways, or even whole genomes (e.g. Nevers et al., 2017). Based on such studies, it has been shown that some protein regions are more conserved than others, depending on structural and functional constraints. For example, it is widely accepted that functionally less important parts of a molecule evolve faster than more important ones.
Numerous approaches have been developed to identify the conserved regions in protein families, e.g. (Livingstone and Barton, 1993; Lichtarge et al., 1996), among many others. These methods are often focused on the identification of structural/functional domains, with domain level resources such as those integrated in InterPro (Finn et al., 2017). Some methods have also been developed to identify conserved motifs in proteins, notably eukaryotic linear motifs (Dinkel et al., 2016), or even single residues (Chakraborty and Chakrabarti, 2015). A limitation of these approaches is that they generally identify conserved elements (domains/motifs/residues) at the protein family level. Such conservation may indicate sites of central and common importance to the protein’s structure or function. The identification of conserved elements within specific sub-families has been less well developed, although some methods do exist for such sub-family level studies, notably for the identification of individual sites in the sequences of related proteins that might lead to functional variation, otherwise known as specificity-determining sites or specificity-determining positions. We focused our work on the identification of conserved elements within sub-families that may suggest regions (domains/motifs) of functional specialization across sub-families of related proteins, and that are essential to understand the function and/or evolution of a protein family.
2 Implementation
We developed PROBE to study the evolution of protein families at the block level. PROBE first clusters the sequences in an MSA into sub-families automatically. This is currently done using Secator (Wicker et al., 2001), but other sequence clustering methods could be used. Then, blocks are identified that are conserved either in the complete alignment or within specific sub-families, using the Bayesian approach we developed previously (Vanhoutreve et al., 2016). For visualization, the blocks are mapped onto a phylogenetic tree that can be constructed automatically or can be input by the user. The blocks are organized in a matrix layout, where each row of the matrix represents a sequence and each column represents a conserved block. A color is assigned arbitrarily to each column (block) to facilitate the visualization. The same colors are used to display the blocks on the multiple sequence alignment.
The PROBE website allows the user to upload either a multiple sequence alignment in FASTA format, in which case conserved blocks will be calculated automatically, or the previously saved results of a PROBE study. After calculation, the results can be downloaded in XML format (MACSIMS; Thompson et al., 2006) or can be visualized in the browser (tested in FireFox, Chrome and Safari). A modified version of jsphylosvg (Smits and Ouverney, 2010) is used to display the phylogenetic tree and the blocks matrix. A modified version of MSAViewer (Yachdav et al., 2016) is then used for the graphical display of the blocks on the MSA.
3 Example
Figure 1 shows an example analysis of an alignment of ENTH (epsin N-terminal homology) domain containing proteins, with similarity to human tepsin protein (AP4AT_HUMAN). Tepsin associates with the adapter-like complex 4 (AP-4) involved in vesicular trafficking of proteins at the trans-Golgi network. It is encoded by a protein of 525 amino acids having an N-terminal ENTH domain, an intervening unstructured segment, a central VHS (Vps27, Hrs, Stam)/ENTH-like domain and a C-terminal unstructured segment. These two domains are annotated for all the sequences in Interpro (respectively, IPR013809 and IPR035802), and correspond to PROBE blocks conserved at the family level. Human tepsin is also known to contain two short linear motifs (defined in Fig. 1C) in its C-terminus that interact with the AP-4 complex (Mattera et al., 2015). From the PROBE display, it can be seen that the first motif, which interacts with the β4 sub-unit of AP-4 ([GS]LFXG[ML]X[LV] in Fig. 1B and C), is conserved in all three groups (animals, stramenopiles and green plants), while the second motif, interacting with the ϵ sub-unit (S[AV]F[SA]FLN in Fig. 1B and C), is restricted to animals, in agreement with the experimental results.
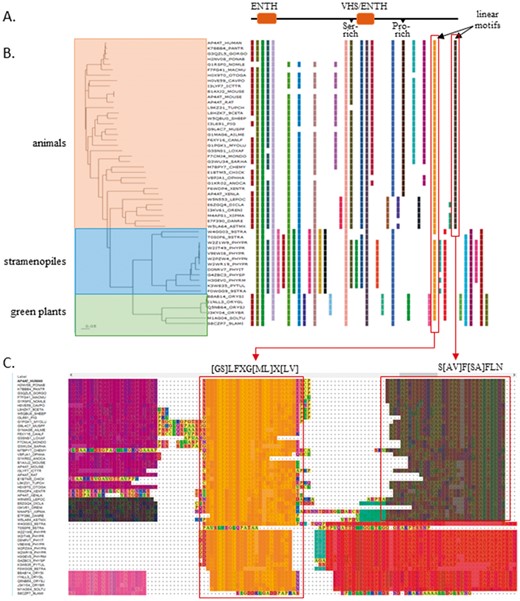
Tepsin family proteins. (A) Domain structure and conserved motifs in human tepsin protein (AP4AT_HUMAN). (B) Phylogenetic tree annotated with family and sub-family level blocks identified by PROBE. (C) Blocks mapped onto the multiple sequence alignment
In conclusion, PROBE provides a fully automatic analysis of protein family conservation, and can be used to identify important functional features in complex multi-domain or multi-functional protein families. Only the MSA is required as input, as sub-families are identified automatically and conserved blocks are identified without using a priori information about known domains/motifs. However, it is also clear that the biological significance of the sequence blocks can only be interpreted by further integrating structural/functional information. A number of limitations remain, in particular concerning the detection of duplicated and/or rearranged blocks. In combination with analyses at the single site level, PROBE should facilitate studies of sequence–structure–function-evolution relationships and opens the way for the construction of phylogenetic profiles at the protein block level.
Acknowledgement
The authors thank the BISTRO and BICS Bioinformatics Platforms for their help.
Funding
This work was supported by the Agence Nationale de la Recherche (BIPBIP: ANR-10-BINF-03-02; ReNaBi-IFB: ANR-11-INBS-0013), and Institute funds from the CNRS, and the Université de Strasbourg.
Conflict of Interest: none declared.
References


{kind=link}