





Abstract
The budding yeast Saccharomyces cerevisiae is a model species powerful for studying the recombination of eukaryotes. Although many recombination studies have been performed for this species by experimental methods, the population genomic study based on bioinformatics analyses is urgently needed to greatly increase the range and accuracy of recombination detection. Here, we carry out the population genomic analysis of recombination in S.cerevisiae to reveal the potential rules between recombination and evolution in eukaryotes.
By population genomic analysis, we discover significantly more and longer recombination events in clinical strains, which indicates that adverse environmental conditions create an obviously wider range of genetic combination in response to the selective pressure. Based on the analysis of recombinational double strand breaks (DSBs)-intersected genes (RDIGs), we find that RDIGs significantly converge on specific disease- and adaptability-related pathways, indicating that recombination plays a biologically key role in the repair of DSBs related to diseases and environmental adaptability, especially the human neurological disorders. By evolutionary analysis of RDIGs, we find that the RDIGs highly prevailing in populations of yeast tend to be more evolutionarily conserved, indicating the accurate repair of DSBs in these RDIGs is critical to ensure the eukaryotic survival or fitness.
Supplementary data are available at Bioinformatics online.
1 Introduction
The budding yeast Saccharomyces cerevisiae has long served as an important model organism for higher eukaryotes as it could promote the combination of classical genetics and biochemistry with recombinant genetics (Amberg et al., 2016), and its cells have striking similarities to mammalian cells at both the organelle and macromolecular level (Braconi et al., 2011). Just recently, a systematic study has shown that most of the genes conserved between yeast and human play the same exact roles in both organisms, and human genes can completely substitute for yeast orthologous genes, raising that humanizing entire cellular processes or pathways in yeast should be feasible (Kachroo et al., 2015). Due to the high biological relevance between yeast and human, biological findings in the budding yeast can be efficiently translated into new insights about human biology (Skrzypek et al., 2018) .
It has been known that genomic instability is an important factor for human diseases. Though endogenous or exogenous genotoxic assaults constantly destroy the genome integrity and lead to DNA damages, cells usually repair them by various mechanisms (Jeggo et al., 2016). Here we focus on the most hazardous damage, namely the double strand breaks (DSBs), which are generally repaired by two main mechanisms, the error-free homologous recombination (HR) and the fast but error-prone non-homologous end-joining (NHEJ), respectively. These mechanisms are shared by eukaryotes (Carroll et al., 2014), but HR is usually less frequently used than NHEJ (Suhane et al., 2015). Considering HR is an accurate DNA repair mechanism, we supposed that recombinational DSBs-intersected genes (RDIGs) were associated with special biological significances.
As the most lethal form of DNA damage, DSBs pose a great potential risk to human health. For example, defects in DNA repair pathways are the causal factors for atherosclerosis, and leaded to higher amounts of DNA damages after exposure to reactive oxygen species (Mahmoudi et al., 2006). When HR repair was defective, smooth muscle cells became increasingly senescent in patients with Hutchinson–Gilford progeria syndrome (Zhang et al., 2014), indicating DNA damages were related to aging (Gorbunova et al., 2007). Differentiation of osteoblasts was inhibited obviously by DNA damages, which contributed to the weakened bone structure in mouse model (Schmidt et al., 2012). These examples suggest that DSBs are associated with human diseases, but it is still unclear which diseases significantly rely on the error-free HR repair mechanism. Hence, we undertake this study aiming at exploring the biological feature and function of RDIGs, which reflects what potential roles the recombination may play in human.
2 Materials and methods
2.1 Strain and data
Based on the phylogenetic relationship previously built by us (Yang et al., 2018), we totally selected 42 strains with consideration of the geographic and environmental origins (Supplementary Table S1), and downloaded the corresponding genome sequences from the NCBI FTP site (ftp://ftp.ncbi.nih.gov/genomes/all/) in August 2016. These 42 strains represent an extensive geographic distribution, and all these genomes are well annotated and highly complete with more than 99% integrity. In order to facilitate the following analyses, each sequence header of every strain genome was unified according to the strain name, chromosome and corresponding identifier. In addition, the protein sequences and GFF3 annotations were also regenerated for each strain based on these gbff files from NCBI by a BioPerl Script.
2.2 Identification of homologous sequences
First, all the genomes were split into individual chromosome sequence files. Then, all the sequences of each chromosome were aligned by Mugsy, which was a robust whole-genome alignment program suitable for accurately aligning the closely related whole-genome sequences (Angiuoli et al., 2011). And then, we extracted all the local aligned blocks of every chromosome, and generated a multi FASTA format file for each of local aligned blocks. However, these blocks with the length <1 kbp or the number of homologous sequences <3 would be viewed as ineligible blocks.
2.3 Phylogenetic network analysis
We adopted these aligned blocks containing 42 homologous sequences to construct the phylogenetic network. First, eligible aligned blocks were realigned separately to produce more accurate alignments by using the program MAFFT v7.271 with default parameters (Katoh et al., 2013). Then, all these realigned blocks were concatenated to a single alignment. And then, all the single nucleotide polymorphism (SNP) loci were extracted to generate the resulting alignment. Finally, this alignment was used as the input of NeighborNet program in SplitsTree v4.14.5 (Huson et al., 2006) to construct the phylogenetic network with default settings. In addition, a statistical analysis was carried out by the pair-wise homoplasy index (PHI) test (Bruen et al., 2006) as implemented by SplitsTree program to estimate the situation of recombination among these strains.
2.4 Detection and analysis of recombination events
After we constructed the phylogenetic network and carried out the PHI statistical test for recombination, we employed seven different methods, the RDP (Martin et al., 2000), GENECONV (Padidam, 1999), 3SEQ (Boni et al., 2007), BOOTSCAN (Salminen et al., 1995), MAXCHI (Smith, 1992), SISCAN (Gibbs et al., 2000) and CHIMAERA (Posada et al., 2001), respectively, to detect the recombination signal, and all these methods were implemented in Recombination Detection Program (RDP4) Beta v. 4.91 (Martin et al., 2015). The breakpoint position of recombination events was determined by RDP4 using a hidden Markov model. In this process, we identified the recombination events separately for each of eligible multiple alignments. And then, all the outputs of RDP4 were parsed to get the simplified and reliable results, but we only considered those recombination events that at least five out of seven methods showed the significant P-value. Finally, we performed the statistical analysis, as well as the association analysis of recombination with environmental factors.
2.5 Identification of recombinational DSBs-intersected genes (RDIGs)
In this study, RDIGs refer to the genes including recombinational DSBs. The identification and functional analysis of RDIGs may be important for exploring the biological significance of recombination because recombinational DSBs were previously reported to be an extraordinarily non-random distribution along the whole genome (Lam et al., 2015). So we identified all the RDIGs and calculated the average frequency of recombinational DSBs by a custom PERL script. In addition, we also examined the distribution of recombinational DSBs in each chromosome by 2 kbp non-overlapping windows, and plotted them by Circos program (Krzywinski et al., 2009).
2.6 Functional enrichment analysis
Here we used the S.cerevisiae strain S288c as reference, and reannotated all its protein-coding genes based on the latest functional data (please see the Supplementary Material for details). Then, a standard procedure EnrichPipeline was adopted to carry out the enrichment analysis of GO term/KEGG pathway (Beissbarth et al., 2004; Huang et al., 2009). In this process, the procedure firstly identified the significantly enriched results according to the P-values by Chi-square test and Fisher exact test. And then, the P-values of gene set enrichment were further adjusted by the default method fdr to filter out those false-positive results. Finally, we took the cutoff of 0.05 for the adjusted P-values as statistical significance.
2.7 Estimation of dN/dS value
In order to estimate the correlation between the evolutionary rate and the frequency of recombinational DSBs, we conducted the dN/dS analysis of orthologous genes as follows. First, orthologous relationships of gene between strains were identified by OrthoMCL software (Li et al., 2003) with the E-value cutoff of 1.0E-05. Then we adopted the best-performing program PRANK (Fletcher et al., 2010; Löytynoja et al., 2005) to generate the codon alignment for each orthologous group without paralogs or missing genes. Finally, the dN, dS and dN/dS were estimated by the maximum likelihood using the codeml program of PAML software package (Yang, 2007).
3 Results
3.1 Phylogenetic network
Originally, we constructed a reticulate phylogenetic network based on the alignment consisted of SNP loci (358 kbp) which were extracted from 240 eligible aligned blocks by a custom Perl script. As shown in Supplementary Figure S1, a complex and reticulate tree topology structure was presented, which indicated the presence of significantly conflicting phylogenetic signals. Generally, conflicting signals were caused by genetic recombination. By the PHI test analysis, we detected the statistically significant evidence of recombination (P = 0.0) in the 42 budding yeast strains. Thus, a more comprehensive assessment of recombination events was necessary for the insight into the underlying biological significance.
3.2 Overview of recombination events
There were totally 762 blocks after filtering the ineligible blocks. By PHI test analysis, we detected the statistically significant evidence of recombination in the 42 budding yeast strains. Then, we identified all possible recombination events (Supplementary Table S2) by the RDP4 program and compared these results predicted by seven different methods implemented in this program, finding that the number of recombination events predicted by different methods ranged from 12 935 to 16 107 (Fig. 1a). According to the overlapping relationships constructed by Venn diagram, we found very few recombination events with significant signals detected by only one method (Fig. 1a), while the number of recombination events with significant signals detected by all seven methods was far greater than the others (Fig. 1b), indicating that these seven methods had a good consistency in the detection of recombination signals. Please note that the two sets of data presented in Figure 1b have a good correspondence. For example, the case with number of methods =5 indicated by red dot includes three cases, namely 5, 6 and 7, indicated by bar plot.
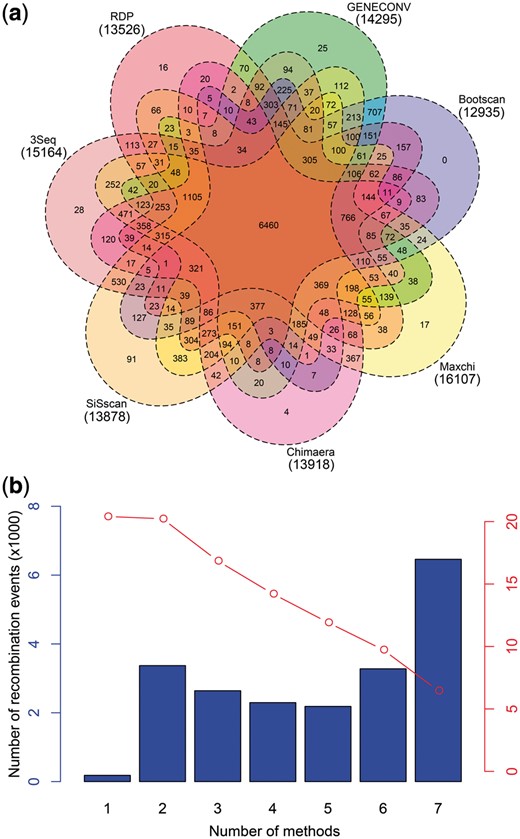
Comparison of the results obtained from seven detection methods of recombination. (a) Venn diagram showing the overlap of recombination events predicted by seven different methods. (b) Bar plot showing the number of recombination events with significant signals detected and only detected by the corresponding number of methods, and red dots linked by red line showing the total number of recombination events with significant signals detected by no less than the corresponding number of methods
Only these recombination events with signals identified to be significant (P-value <1.0E-05) by no less than 5 methods were retained for the following analyses, totaling 11, 921 recombination events. Subsequently, by statistical analysis we found that the average length of recombination events in chromosome 2 were obviously larger than that in others, and the number of recombination events in chromosome 2, 4 and 15 was significantly (P-value =2.9E-03) more than those in others (Supplementary Fig. S2). By linear regression analysis, we found that the number of recombination events was positively correlated to the length of chromosomes with a correlation coefficient of 0.67 (Supplementary Fig. S3a), but the average length of recombination events had almost no correlation to the length of chromosomes with an insignificant correlation coefficient of 0.16 (Supplementary Fig. S3b). Remarkably, in the smallest four chromosomes, the average length of recombination events had an obvious positive correlation to the length of chromosomes, but the number of recombination events had a strong negative correlation to the length of chromosomes, which was in accordance with previous experimental findings on the smallest four chromosomes that the number of recombination events decreased with the length of chromosomes (Gerton et al., 2000). In addition, we also estimated the guanine-cytosine (GC) content of recombination sequences, finding that sequences from recombination events had slightly lower GC contents than those in genomic sequences (Supplementary Fig. S4).
3.3 Source analysis of recombination
In order to investigate the correlations between recombination and geographical or environmental factors, we constructed the network relationship of recombination events, which clearly showed the intricate relationship of recombination among strains from different sources. By this network diagram (Supplementary Fig. S5), we observed that recombination appeared to occur more frequently in clinical strains than non-clinical strains, indicating that these strains in clinical environments suffered from stronger selection pressures, because there always existed a large number of exogenous agents like pharmaceuticals and ionizing radiation. In order to assess whether the number and length of recombination events in clinical environments differed significantly from those in other environments, we carried out the Wilcoxon test to detect the statistical significance of difference between any two environments. As a result, we found the number of recombination events in clinical environments was obviously more than that in the other two environments (wine and other, respectively) (Fig. 2a), but the total length of recombination sequences in clinical environments was significantly longer than that in either (Fig. 2b). These findings indicated that strains from harmful environments had significantly more genetic combination than those from benign environments, which was in accordance with what had been observed in the fungus Arthrobotrys oligospora (Zhang et al., 2013).
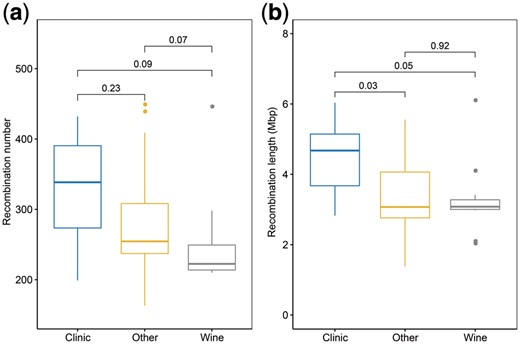
Relationship between environmental factors and recombination. (a) Comparison of recombination number between any two environmental factors. (b) Comparison of recombination length between any two environmental factors. Significance of above two analysis is estimated by the pairwise Wilcoxon test. Clinic, wine and other refer to the clinical, wine and other environments respectively
3.4 DSBs and RDIGs
Based on all the recombination events detected above, we identified 23 356 recombinational DSBs from genomes of 42 budding yeast strains, and plotted the distribution of DSBs along each chromosome. As a result, we found that DSBs were obviously distributed non-randomly along chromosomes (Fig. 3), which was consistent with previous findings (Székvölgyi et al., 2015). Then, we mapped the positions of all these DSBs to the genome of strain s288c, and totally identified 3060 RDIGs. Actually, RDIGs were such genes spliced together by different portions of two homologous genes, therefore they also belonged to the mosaic genes. We calculated the average frequency of recombinational DSBs occurring in RDIGs by a custom Perl script. We focused more on these RDIGs with high frequency (≥0.9) of recombinational DSBs (referred as HRDIGs here), because these genes were highly dependent on error-free HR. Here, we totally identified 60 HRDIGs. Considering that perfect complementary pairings of invasive strand with template were crucial for the initiation of HR (Maher et al., 2011), we speculated that HRDIGs were likely to be evolutionarily conserved and biologically significant genes.
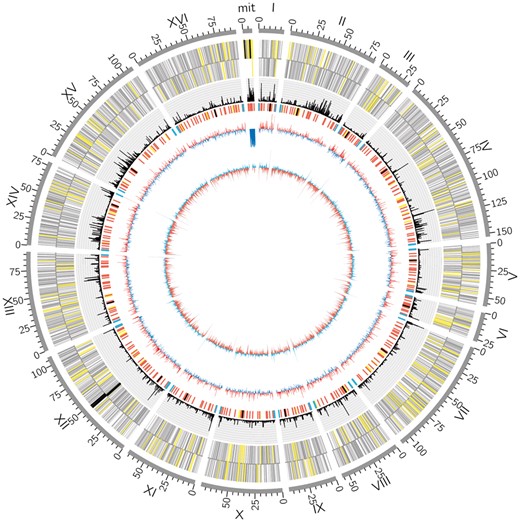
Distribution of DSBs along the genome of strain S288c. From outer to inner, the circle represents the genome coordinate (grey), all the genes located in forward and reverse strands (yellow, black and grey indicate the tRNA, rRNA and other genes, respectively), the percentage of DSBs (black), some important sequence elements (black, red, blue, yellow and green indicate the centromere, replication origin, telomere, mobile element and repeat region, respectively), GC content (blue/red) and GC skew (blue/red), respectively
3.5 Results of KEGG enrichment analysis in HRDIGs
By KEGG enrichment analysis, we found 13 pathways related to human diseases significantly enriched in HRDIGs (Fig. 4; Supplementary Table S3). Specifically, there were three neurological disorders (NDs) (PD, HD and AD), which indicated that organisms relied on HR to thwart the occurrence of NDs. This was supported by previous findings that accumulation of DNA lesions was tightly related to NDs (Maynard et al., 2015), and organisms may resist not only the normal aging but also age-related NDs by reliable DNA repair mechanism (Leandro et al., 2015). And there were two cancer pathways (prostate cancer and chemical carcinogenesis, respectively), which was in accordance with previous findings that inaccurate repair of DSBs was related to the development of certain cancer (Dexheimer, 2013), and defects of recombination predisposed to cancer (Martino et al., 2016). There were also three other disease pathways, the non-alcoholic fatty liver disease, measles, fluid shear stress and atherosclerosis, respectively. Indeed, growing evidence pointed to the association of DNA damages with metabolic syndromes, such as non-alcoholic fatty liver disease and atherosclerosis (Gray et al., 2011; Jackson et al., 2009). As to the enrichment of measles pathway, previous experimental study observed a similar result that all the measles cases had an obviously increased frequency of DSBs (Koskull et al., 1977). Moreover, there were also five disease-related pathways, namely the IL-17 signaling pathway, oxidative phosphorylation, antigen processing and presentation, Th17 cell differentiation and cardiac muscle contraction, respectively. These results together with the results of KOG and GO analysis (Supplementary Figs S6 and S7 and Table S4) suggested that HRDIGs were tightly associated with certain human diseases. Moreover, there were also some pathways related to specific adaptability. For instance, estrogen signaling pathway regulated a plethora of physiological processes in mammals (including cardiovascular protection, reproduction and behavior) (Rotroff et al., 2013), the drug metabolism and metabolism of xenobiotics by cytochrome P450 were responsible for xenobiotics biodegradation and metabolism (Kirchmair et al., 2015), and the two-component system acted as a basic stimulus-response mechanism to permit organisms to sense and respond to the changes of environmental conditions (Kreamer et al., 2015). These results indicated that functional impairment of HRDIGs was likely to result in obvious decrease of environmental adaptability, hence DSBs occurring in HRDIGs were highly dependent on the error-free HR repair mechanism.
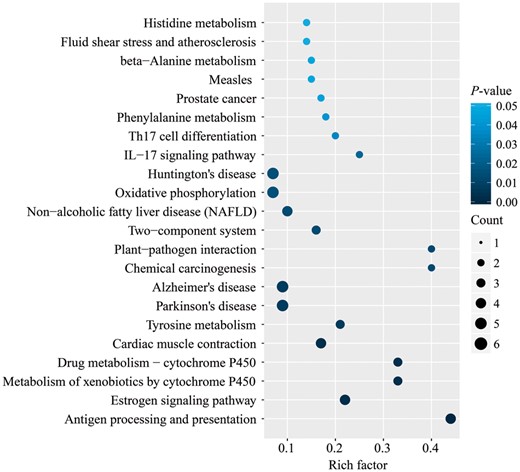
Scatterplot for significantly enriched KEGG pathways in HRDIGs. Rich factor is the ratio of the number of HRDIGs annotated in this pathway to the number of all genes annotated in this pathway. P-value is the corrected P-value ranging from 0 to 0.05, with a lower value meaning a more significant enrichment. Count represents the number of HRDIGs
3.6 DSBs frequency and dN/dS of RDIGs
By analyzing the relationship of recombinational DSBs frequency and dN/dS value in RDIGs, we found that those RDIGs prevailing more widely in populations tended to keep a lower dN/dS value, and almost all of positively selective genes (PSGs) held an extremely low frequency of DSBs (Fig. 5a). This was consistent with previous studies that high genetic similarity corresponded more significantly to the distribution of DSBs (Magiorkinis et al., 2003). In HR repair, a long stretch of single strand DNA would be generated by the end resection of DSBs (Truong et al., 2013), and this single strand DNA might be the most critical factor for determining if a homolog should be used as the recombination partner (Joshi et al., 2015). In this analysis, we found that the frequency of recombinational DSBs in PSGs was significantly lower than that in negatively selective genes (NSGs) (Fig. 5b), and none of HRDIGs were PSGs. Hence, we speculated that HR preferred to repair these DNA damages in highly homologous regions, which in turn contributed to higher evolutionary conservation in these regions.
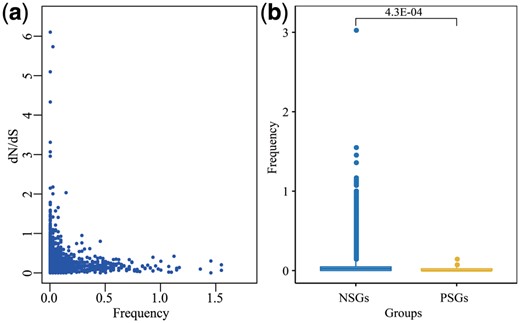
Relationship between recombinational DSBs and genetic evolution. (a) Correlation between recombinational DSB frequency and dN/dS value. (b) Comparison of recombinational DSBs frequency between NSGs and PSGs. Statistical significance analysis is performed by the pairwise Wilcoxon test
4 Discussions
As discussed above, HR is a biologically significant mechanism of repairing DSBs that occur spontaneously or are induced by biological, chemical or physical agents. In the HR process, 5′ end of DSBs is firstly resected to generate an ssDNA tail of ∼800 nt. And then, this ssDNA tail initiates the homologous pairing after invasion, and serves as the primer for DNA biosynthesis. Ultimately, an intact homologous duplex is used as template to repair the broken strands. However, HR is only responsible for a small proportion of DSBs (Sundararajan et al., 2016). Its operating frequency is usually regulated homeostatically, and does not change with alteration of DSB frequency (Modliszewski et al., 2017). Though HR is an error-free DNA repair mechanism, it is also a non-random and biased process. While HR was found to prefer to repair these DSBs in evolutionarily conserved regions as observed above (Fig. 5), there may be some other factors influencing this priority of HR. We supposed that HR would be used preferentially to repair those biologically significant DSBs.
In this study, we found that the number of recombination events had an obviously positive correlation with the length of chromosomes, and also found that the total length of recombination events in clinical strains were significantly longer than that in wine or other strains. A reasonable interpretation for this is that clinical strains are subjected to numerous DNA damages, and need to break up the undesirable combinations and create favorable allelic ones in a wider range. Generally, recombination is essential for restoring the damaged genes, but its DSBs-intersected genes are structured into mosaic genes. By this way, widely prevailing RDIGs in populations generally evolve only with a very low rate (Fig. 5a) so as to make sure that only these highly advantage mutations are accumulated. Hence, RDIGs should have special biological significance, especially HRDIGs that hold high prevalence in the natural populations. As we presumed, HRDIGs are significantly enriched in some critical biological processes and pathways related to the specific adaptability and human diseases, such as NDs, cancers and drug metabolism. All these findings indicate that the HR repair of DSBs may well be crucial for the fitness of eukaryotic organisms, and also contribute to the development of new therapeutic strategies for specific human diseases.
Acknowledgements
The authors would like to thank Prof. Chun-Ting Zhang for the invaluable assistance and inspiring discussions.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31571358, 21621004, 31171238, 11626250 and 91746119).
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}