





Abstract
Pairwise sequence alignment is undoubtedly a central tool in many bioinformatics analyses. In this paper, we present a generically accelerated module for pairwise sequence alignments applicable for a broad range of applications. In our module, we unified the standard dynamic programming kernel used for pairwise sequence alignments and extended it with a generalized inter-sequence vectorization layout, such that many alignments can be computed simultaneously by exploiting SIMD (single instruction multiple data) instructions of modern processors. We then extended the module by adding two layers of thread-level parallelization, where we (a) distribute many independent alignments on multiple threads and (b) inherently parallelize a single alignment computation using a work stealing approach producing a dynamic wavefront progressing along the minor diagonal.
We evaluated our alignment vectorization and parallelization on different processors, including the newest Intel® Xeon® (Skylake) and Intel® Xeon PhiTM (KNL) processors, and use cases. The instruction set AVX512-BW (Byte and Word), available on Skylake processors, can genuinely improve the performance of vectorized alignments. We could run single alignments 1600 times faster on the Xeon PhiTM and 1400 times faster on the Xeon® than executing them with our previous sequential alignment module.
The module is programmed in C++ using the SeqAn (Reinert et al., 2017) library and distributed with version 2.4 under the BSD license. We support SSE4, AVX2, AVX512 instructions and included UME: SIMD, a SIMD-instruction wrapper library, to extend our module for further instruction sets. We thoroughly test all alignment components with all major C++ compilers on various platforms.
Supplementary data are available at Bioinformatics online.
1 Introduction
Aligning biological sequences is among the most prominent algorithmic components in bioinformatics pipelines and tools. It is part of various applications in genomics, such as adaptor trimming (Roehr et al., 2017), read mapping (Langmead and Salzberg, 2012; Li, 2013; Siragusa et al., 2013; Weese et al., 2012), genome assembly (Holtgrewe et al., 2015; Li et al., 2012b), variant detection (Emde et al., 2012; Rausch et al., 2012), local alignment (Kehr et al., 2011) as well as multiple sequence alignments (Notredame et al., 2000; Rausch et al., 2008) and in proteomics, e.g. protein database search (Buchfink et al., 2015; Hauswedell et al., 2014; Ye et al., 2011) and many more. This shows that there is a large range of applications for the alignment problem which differ in several aspects [scoring schemes (Pearson, 2013), banding conditions (Chao et al., 1992), etc.] but all share in the core the same algorithm.
In its standard form, the algorithm was already described in Needleman and Wunsch (1970). It is based on dynamic programming (DP) and computes the sequence alignment for a given pair of sequences for the so-called linear gap costs. Gotoh (1990) refined the DP algorithm to allow affine gap costs. The algorithm consists of an initialization step and a recursion followed by a traceback to obtain the final alignment.
In the following description, we will concentrate on the most common variant, the case of affine gap costs. Given two sequences s1 and s2, with and over an alphabet Σ, a score function , which compares two characters a and b, with and gap penalties ωo for opening a gap and ωe for extending a gap. A sequence of length 0 is called the empty sequence and denoted as ϵ. Further, let be the ith character of a sequence s. In this work we assume zero-based indices when working with sequences. A global alignment between s1 and s2 using affine gap costs can be then computed as follows:
The best alignment can then be determined by inspecting the lower, rightmost cell of M, i.e. . The total runtime of the algorithm is , while the score, without the traceback, can be computed in space.
Given the many implementation variants, it would be prudent to have a common core computation in a library that (a) can be the basis for the various versions of pairwise alignment and (b) which can be parallelized. We developed SeqAn (Döring et al., 2008; Reinert and Gogol-Döring, 2009; Reinert et al., 2017), a general purpose sequence library written in C++ containing many efficient and generic algorithms and data structures for various sequence analysis tasks, e.g. alignments, online searches, indices and offline searches, etc. SeqAn’s alignment module implements among the standard DP algorithms many efficient variants of the aforementioned DP problem. Table 1 assembles an overview over the rich feature set of SeqAn’s alignment module.
The different configuration options for our pairwise sequence alignment module
| Algorithm | Global | Local | Free-end gaps | Banded global | Banded local | Banded free-end gaps |
|---|---|---|---|---|---|---|
| Gap function | Linear gaps | Affine gaps | Dynamic gaps | |||
| Traceback | Score only | Single best | All best | Right gap projection | Left gap projection | |
| Specialized | Split breakpoint | Banded chain alignment | Affine X-drop | Hirschberg | Myers’ bitvector | Myers’ Hirschberg |
| Algorithm | Global | Local | Free-end gaps | Banded global | Banded local | Banded free-end gaps |
|---|---|---|---|---|---|---|
| Gap function | Linear gaps | Affine gaps | Dynamic gaps | |||
| Traceback | Score only | Single best | All best | Right gap projection | Left gap projection | |
| Specialized | Split breakpoint | Banded chain alignment | Affine X-drop | Hirschberg | Myers’ bitvector | Myers’ Hirschberg |
Note: We implemented the standard global and local alignment and a free end-gap version, where every border of the DP matrix can be configured individually for free end-gaps, as well as the banded version of all of them. Our current implementation supports three different gap functions and five traceback options reporting only the score (the traceback is completely disabled), one of the traces of the optimal solution or all of them, while for the latter option an ambiguous gaps placement can be resolved by either a left or right projection (see Supplementary Fig. S2). All these options can be combined arbitrarily creating an exceptionally comprehensive repertoire of DP algorithms containing more than 500 variants. The fourth row shows some special DP implementations available in SeqAn. Most of them are implemented by utilizing our unified DP core (see Section 1 in the Supplementary Material)
The different configuration options for our pairwise sequence alignment module
| Algorithm | Global | Local | Free-end gaps | Banded global | Banded local | Banded free-end gaps |
|---|---|---|---|---|---|---|
| Gap function | Linear gaps | Affine gaps | Dynamic gaps | |||
| Traceback | Score only | Single best | All best | Right gap projection | Left gap projection | |
| Specialized | Split breakpoint | Banded chain alignment | Affine X-drop | Hirschberg | Myers’ bitvector | Myers’ Hirschberg |
| Algorithm | Global | Local | Free-end gaps | Banded global | Banded local | Banded free-end gaps |
|---|---|---|---|---|---|---|
| Gap function | Linear gaps | Affine gaps | Dynamic gaps | |||
| Traceback | Score only | Single best | All best | Right gap projection | Left gap projection | |
| Specialized | Split breakpoint | Banded chain alignment | Affine X-drop | Hirschberg | Myers’ bitvector | Myers’ Hirschberg |
Note: We implemented the standard global and local alignment and a free end-gap version, where every border of the DP matrix can be configured individually for free end-gaps, as well as the banded version of all of them. Our current implementation supports three different gap functions and five traceback options reporting only the score (the traceback is completely disabled), one of the traces of the optimal solution or all of them, while for the latter option an ambiguous gaps placement can be resolved by either a left or right projection (see Supplementary Fig. S2). All these options can be combined arbitrarily creating an exceptionally comprehensive repertoire of DP algorithms containing more than 500 variants. The fourth row shows some special DP implementations available in SeqAn. Most of them are implemented by utilizing our unified DP core (see Section 1 in the Supplementary Material)
This generic implementation makes SeqAn’s alignment module uniquely versatile and ready for use in many of the above-mentioned applications. However, the quadratic run time of the DP algorithms can quickly become the bottleneck in processing large datasets. Thus, in the past decade it was of great interest to accelerate the standard DP algorithms on modern hardware or to run them on HPC (high-performance computing) environments.
Different techniques have been developed and optimized over time to accelerate the DP algorithm on various platforms. In general we differentiate between the two levels of parallelism: thread level, and vector level. Thread-level parallelization refers to the procedure of splitting a process into multiple threads and concurrently executing them on multiple cores on one processor. Vector-level parallelization is the process of simultaneously executing a single instruction on multiple data (SIMD) using dedicated register instructions, also called SIMD instructions. Here the data are packed into extended registers of sizes up to 512 bit such that special arithmetical, logical, bit or other operations can be applied on the data in parallel (Intel, 2016; Jeffers et al., 2016).
The complexity of both approaches for the DP problem depend on the underlying execution layout. For sequence alignments there are two main execution layouts: The inter-sequence and the intra-sequence layout. The former can be used when many sequence alignments can be computed. The problem is then trivially parallelizable, since there are typically no dependencies between the different alignment instances. The latter focuses on accelerating a single-alignment computation and has to deal with the data dependency that originates from the recursion shown in Figure 1. Interestingly, both layouts can benefit from thread-level and vector-level parallelization.
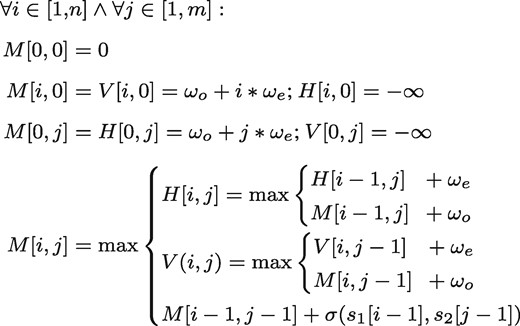
Pseudocode for computing a pairwise sequence alignment with affine gap costs
1.1 Previous work
Much effort has been put into exploring optimal vectorization strategies for alignment algorithms. Several intra-sequence execution layouts of the Smith-Waterman algorithm (local alignment) were investigated, vectorizing either over the minor diagonals of the alignment matrix (Wozniak, 1997), or along the query sequence by means of a sequential (Rognes and Seeberg, 2000) or striped (Farrar, 2007) vector pattern. As an alternative, the inter-sequence layout aligns a single query sequence against a vector of subject sequences (Alpern et al., 1995; Rognes, 2011) making each alignment computation independent and thus achieving potentially higher speedups than the intra-sequence scheme (Daily, 2016; Rucci et al., 2017).
Thread-level parallelization for the inter-sequence layout can be applied if many alignments need to be computed, which is a common use case in bioinformatics pipelines. The workload can be easily partitioned in chunks and then computed concurrently on the different cores of the multi-core processor, manycore processor or accelerator (Blazewicz et al., 2011; Daily, 2016; Rognes, 2011). For the thread-level, intra-sequence layout, strategies have been implemented that are similar to the intra-sequence vectorization layout, including a wavefront-based model progressing along the minor diagonal (Edmiston et al., 1988; Liu et al., 2001) a striped (Li et al., 2012a) or a sequential layout (Khajeh-Saeed et al., 2010).
Most of the work focussing on vector-level and thread-level parallelization for alignment algorithms was done in the context of accelerating the Smith-Waterman kernel for either protein database searches or pairwise sequence alignments of long input sequences on either CPUs with SIMD vectorization (Farrar, 2007; Rognes, 2011; Rognes and Seeberg, 2000), GPUs (Khajeh-Saeed et al., 2010; Korpar and Šikić, 2013; Li et al., 2012a; Liu et al., 2013; Sandes and de Melo, 2013), cell broadband engines (Sarje and Aluru, 2008; Szalkowski et al., 2008) or on more recent architectures like the Xeon PhiTM from Intel® (Liu and Schmidt, 2014; Liu et al., 2014; Rucci et al., 2017). However, the algorithmic components are hard to reuse since they are hidden within these applications and many tools work with outdated instruction sets. Hence, it is crucial for the bioinformatics community to expose these algorithms as a reusable library, which is well maintained, offers an user-friendly and stable application programming interface (API), and supports various kinds of target applications.
The SSW library (Zhao et al., 2013) was one of the first general-purpose libraries that implemented the striped vectorization layout by Farrar (2007) for local alignments only. Recently, the Parasail library was published, which implements several different strategies for the intra-sequence vectorization layout and also extended them for the global and semi-global case (Daily, 2016). Libssa (Frielingsdorf, 2015) and Opal, formerly SWIMD (Šošić, 2014), are two software libraries that implement an inter-sequence vectorization layout following the approach of Rognes (2011). They both offer local and global alignment computations. Opal also implements two variations of the semi-global alignment with different free end-gap settings. Libssa, similar to SSW, computes also a trace of the optimal alignment after the highest scoring subject sequence has been identified. All mentioned libraries support SIMD instructions for SSE4. Parasail, Opal and Libssa also implement the AVX2 instructions and only Parasail additionally supports a first version of 512 bit instructions, known as Intel® IMCI (initial many core instructions) only available for the first generation of Xeon PhiTM coprocessors (KNC), and AltiVec instructions. All libraries support an additional integer saturation mode, which executes the alignment with the smallest possible bit range and reruns alignments with a higher bit range, if an integer underflow/overflow was detected during the matrix computation. While Parasail and SSW are actively developed, it seems that Opal and Libssa are not further maintained.
1.2 Our contribution
One of our major design goals for SeqAn is the genericity of data structures and algorithms, while still being highly efficient. With this key paradigm in mind, we redesigned and refactored SeqAn’s alignment algorithm such that all different DP variants listed in Table 1 are unified in one central DP kernel. We make use of template meta-programming (Vandevoorde and Josuttis, 2002) and tag dispatching to select the most performant execution models for the chosen alignment configuration at compile time. Thus, we reduce the overhead of redundant kernel implementations, which makes the entire code easy to maintain and to extend, e.g. by adding a new gap function like the affine-like dynamic gap model (Urgese et al., 2014) or by adding a banded version of the algorithms. This versatility is the first main contribution of this work.
In addition, we extended the DP kernel to generically support an inter-sequence vectorization layout (see Fig. 2). While existing methods only support one-versus-many alignments (Frielingsdorf, 2015; Rognes, 2011; Šošić, 2014), we generalized the layout to also support many-versus-many alignments. By implementing a templatized vector alphabet type, which wraps either SSE4, AVX2 or AVX512 instructions we were able to inherently vectorize almost all of the DP features mentioned in Table 1. Furthermore, we implemented a wrapper for the UME::SIMD library (Karpiński and McDonald, 2017), which allows our alignment kernels to be accelerated with the IMCI (https://software.intel.com/en-us/node/694272, accessed November 13, 2017), NEON (ARM, 2007) or AltiVec (Freescale Semiconductor, 1999) instruction sets too.
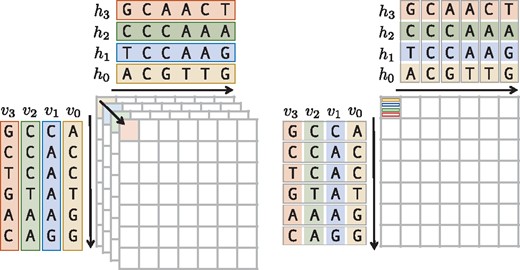
Inter-sequence vectorization. The sequences on the left side (array-of-structures layout) are converted into an array of SIMD vectors on the right side (structure-of-arrays layout), i.e. the number of matrix calculations is reduced from four to one by computing four elements packed into one SIMD-vector simultaneously
On top of our vectorized DP kernel, we implemented two layers of thread-level parallelization, which can be selected by the user. The first follows the inter-sequence layout, which assumes that many independent alignments can be computed, favorably with equal-sized sequences, which is usually the case when working with reads from various sequencing technologies (Metzker, 2010). The second implements an intra-sequence layout based on a dynamic wavefront model using a dependency graph on sub-alignments in combination with a work stealing algorithm (Blumofe and Leiserson, 1999) rather than parallelizing the for-loop over the minor diagonals (Edmiston et al., 1988; Liu and Schmidt, 2014; Liu et al., 2001, 2014). We then combined the vectorized DP kernel with the thread-level parallelization to multiply the performance gains from both acceleration methods.
Finally, we extended the wavefront model by a generic alignment scheduler in order to asynchronously process many alignments of arbitrary size. Conceiving and implementing these features as part of SeqAn’s DP unit creates a truly generic intra- and inter-sequence accelerated alignment module that runs on any general-purpose multi-core CPU as well as on manycore processors such as the new Xeon PhiTM (see Fig. 3).
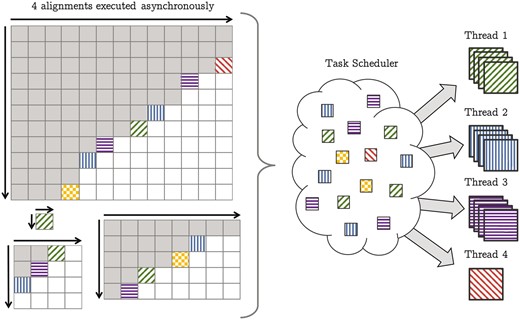
Generalized wavefront model to compute multiple pairwise sequence alignments split in many sub-alignments. The gray tiles are already computed. The colored (filled) tiles are ready for execution and wait in the task scheduler for the next available thread. If a thread becomes available it tries to pick l = 4 many sub-alignments from the scheduler and computes them vectorized. In this example this applies to thread 1, 2 and 3. The sub-alignments can come from different alignments, e.g. the blue tiles (vertical bars) currently processed by thread 2 originate from three different alignment instances. If not enough tiles are available the thread picks solely a single tile. Here thread 4 could grab at most three tiles and therefor continues in the scalar mode computing just one sub-alignment. Also there is no limitation on the size of the sequences to be aligned, such that a single alignment can consist of just one tile
In this paper, we will describe the design of the wavefront method as well as its adaption to vectorized DP kernel and the generic alignment scheduler. We will then compare our implementation to Parasail for three different use cases using the newest Intel® processor Skylake.
In addition, we provide a detailed description of the implementation of the unified generic DP kernel and which design strategies were realized to achieve high efficiency in the Supplementary Material. Further, we will explain the adaptions made to vectorized and parallelized DP implementation based on the unified DP kernel. Then evaluate the different alignment parallelization modes on different use cases using three CPU architectures. We will then investigate the three use cases from this paper with respect to three different processor architectures and its implications for SeqAn’s alignment library.
In general, we will show that our methods outperform existing strategies for all applied use cases on all tested platforms by factors from 3 to 17 and that we could speedup alignment computations up to a factor of 1600 compared with a sequential scalar execution. We further show first results for a vectorized sequence alignment using the AVX512-BW instruction, which extends the AVX512 instruction set with byte and word-packed integer operations (Intel, 2016).
2 Materials and methods
In the subsequent sections, we will describe our generic vector- and thread-level parallelization for the alignment module, where we take advantage of the generic design of the DP kernel.
2.1 Vector-level parallelization
In our vectorization scheme, we follow the inter-sequence layout, in which many alignments are executed in one vector unit concurrently. Opposed to known implementations (Rognes, 2011), we generalized the inter-sequence vectorization layout, to calculate alignments in a many-to-many fashion, i.e. we allow multiple query sequences to be aligned against multiple subject sequences packed in one vector.
Figure 2 shows this layout in a toy example, where sequence hi is aligned against sequence vi for . Here, we compute in a single pass over the alignment matrix 4 alignments using SIMD instructions. The vectorized DP kernel is entirely implemented by means of the generically unified DP core, allowing us to vectorize most of the variants listed in Table 1. In Section 2 of the Supplementary Material, we give a more detailed description of the adaptions we made to the original version of the unified DP core.
2.2 Thread-level parallelization
To target a maximal number of bioinformatic domains, we implemented two parallelization schemes, following the inter-sequence and the intra-sequence layout. The former assumes a set of pairwise sequence alignments to be computed. In this case, the set of pairwise alignments will be split into subsets that can be computed concurrently on multiple cores. The latter approach follows the wavefront parallelization model, where a single alignment is parallelized along its minor diagonal (Liu et al., 2014). However, in contrast to current implementations, we use a more generic framework that utilizes a dependency graph in combination with a work stealing approach to generate a dynamic wavefront model.
2.2.1 Dynamic wavefront parallelization
Similar to Liu et al., we split the DP matrix into tiles of size , where β is an user configurable constant, producing many sub-alignments. There are many tiles in horizontal direction and tiles in vertical direction. We then generate a directed acyclic dependency graph with V being the set of nodes, where each maps to exactly one tile , with and and E being the set of directed edges connecting those tiles in G (see Supplementary Fig. S3). In particular, we add an outgoing edge for every node v, such that every tile is connected with its successors and . Except for the nodes mapping to and , which have an out-degree of 1, respectively 0, all nodes v are connected to exactly two successor nodes. We assign every node v a dependency count initialized to its corresponding in-degree.
To put it differently, the mapped tile of node v is scheduled for execution if all its predecessors have finished their computation, i.e. v’s dependency count equals 0. We implemented a thread-safe task scheduler, which allows threads to add and pull tasks from the scheduler concurrently. The parallel execution of the alignment is triggered when the source mapping to is scheduled for execution by the parent thread. Due to a work stealing mechanism the execution of the tiles follows a dynamic wavefront progressing from the source toward the sink of the dependency-graph (see Fig. 3). A condition variable is used to broadcast that the computation of the sink () has completed. The parent thread waits until it gets notified by its associated condition variable and passes the result to a user-definable callback function.
Two matrix wide buffers, i.e. for the horizontal dimension (bufh), and the vertical dimension (bufv) respectively, are used to synchronize the state of the alignment cells with the successor nodes. Thereby, the execution in the dependency graph guarantees that there is no race condition on the buffer values, as it is not possible that two tiles in the same column or same row of the dependency graph are executed concurrently by different threads. Supplementary Figure S4 depicts the partitioning of the DP matrix and how the buffers are accessed during the wavefront execution.
2.2.2 Integrating inter-sequence vectorization
To gain substantial speed-ups, we take advantage of our tiling strategy by gathering scheduled sub-alignments, which are by definition independent and align them using our generalized inter-sequence vectorization layout described in Section 2.1. Therefore, the current thread extracts l many sub-alignments from the task scheduler, with l being the number of packed alignments per SIMD vector, and executes them with our generically vectorized many-to-many alignment core (threads 1, 2, 3 in Fig. 3). If less than l tasks are available, the current thread extracts only one thread and computes it using the scalar DP kernel, e.g. thread 4 in Figure 3. Thus, the workload is dynamically split between scalar and vectorized alignments to efficiently deal with the beginning and the end of the matrix where there might be not enough sub-alignments available for a vectorized execution.
2.2.3 Generalized alignment scheduler
We implemented an asynchronous alignment scheduler that maintains a set of processed alignments using a pool of helper threads. Each helper thread is responsible for the preparation and management of a single alignment, i.e., generating the dependency graph, setting up the tile buffers, extracting the solution from the thread-local storages and continuing the process by invoking the user-defined callback function. The number of helper threads can be configured at runtime and can be larger than the number of worker threads that do the actual alignment computation. Thus, given many alignments, it is possible to add them dynamically and to produce enough work for the wavefront model, such that most alignments are computed in vectorized mode, resulting in a highly scalable and extremely flexible alignment execution framework. Figure 3 shows a simplified version of four different alignments computed concurrently with the generalized alignment scheduler.
2.2.4 Execution policies
Providing a flexible and user-friendly interface is a crucial concern of SeqAn’s library design. To satisfy these requirements we implemented an execution policy as the central data structure to configure the options for the different parallelization strategies. The user can choose between three modes for the thread-level parallelization: sequenced, chunked and wavefront mode. The sequenced mode executes the alignment without parallelization. The chunked policy splits a set of alignments into chunks of equal size and executes the chunks concurrently. The wavefront mode uses the above-described dynamic alignment scheduler. In addition, the user can combine any of the thread-level parallelization policies with scalar execution or vector-level parallelization. In the Supplementary Material, Listing S1 demonstrates the efficiency and the usability of the execution policies.
3 Results
We implemented a benchmark application (https://github.com/rrahn/align_bench.git (14 February 2018, date last accessed)) to evaluate the performance of our generic vectorized and parallelized pairwise sequence alignment module. The data for this evaluation is stored on a separate ftp server (ftp://ftp.mi.fu-berlin.de/pub/rmaerker/align_bench/ (14 February 2018, date last accessed)).
We evaluated three common use cases: overlap alignments of short Illumina reads, local alignment of large-scale sequences and semi-global alignment of PacBio reads.
For all benchmarks we used the affine gap model with a score of 6 for match, –4 for mismatch, and –1, respectively –11, for the gap extension and gap open penalties. To be comparable with other software we run all experiments with disabled traceback.
We compared our implementation if applicable with Parasail in version 2.0.2 which was the most recent version available at the time of this evaluation. We also tried available applications based on Libssa and Opal, however, both tools only work for protein sequences and only facilitate the database search problem, such that we had to exclude them from the benchmarks.
All benchmarks were performed on a two socket Intel® Xeon® Gold 6148 CPU (abb. SKX) with 40 physical cores at a base frequency of 2.4 GHz. We tested our algorithms with SSE4, AVX2 and AVX512 instructions. The platform run a centos 7 unix kernel. The Intel® turbo boost feature was disabled. All benchmark applications were compiled with g++-7.2.0. We used the environment variable GOMP_CPU_AFFINITY to pin each thread to the cores in a round-robin fashion if the program used OpenMP threads. In our wavefront model, we generically use native threads using STD’s thread support library and programmatically pinned the threads by accessing the native thread handlers.
In addition, we performed the benchmarks on a Intel® Xeon® E5-2650 V3 CPU (abb. HSW) with 20 cores and a Intel® Xeon PhiTM 7250 CPU (abb. KNL) with 68 cores. A detailed comparison of the benchmarks with respect to the different architectures can be found in Section 3 of the Supplementary Material. In the following we will keep the analysis limited to the SKX results.
3.1 Overlap alignments of Illumina reads
In this use case we computed 12 497 500 pairwise sequence alignments of 150 base long Illumina single-end reads simulated with Mason in version 2.0.8 (Holtgrewe, 2010) using chromosome 10 of GRCH38 as the reference genome. We used the chunked execution policy for this data on all 40 threads using 16 bit score width, which performed best in our benchmarks and also included Parasail results to compare both library implementations. We run SeqAn using global, semi-global and local alignment using the non-banded and the banded algorithm. The band was configured with a bandwidth of 16 (8 bases to either site) to represent an error rate of 5%. We compared our results with the respective alignment mode of Parasail. We tried all possible configurations and chose the one with the best results. Note that Parasail supports SIMD vectorization only up to AVX2, such that there is no data available for AVX512 and it also does not support vectorized banded alignments.
The fastest algorithm was the banded version of SeqAn using AVX512 finishing the computation in 0.24 s (AVX2: 0.4s; SSE4: 0.61s). The timings for the banded local alignment were similar: AVX512: 0.26s; AVX2: 0.42s; SSE4: 0.74s. The banded version was roughly three times faster than the non-banded case for the global alignment (AVX512: 0.68s; AVX2: 1.35s; SSE4: 2.68s) as well as for the local alignment (AVX512: 0.80s; AVX2: 1.55s; SSE4: 3.26s). Compared with Parasail this is 5 up to 17 times faster for the global alignment (AVX2: 4.00s; SSE4: 3.88s) and the local alignment (AVX2: 3.96s; SSE4: 4.11s). In total SeqAn achieves a peak performance of roughly 420 giga cell updates per second (GCUPS), while Parasails peak performance was 77.86 GCUPS. In other words, we can run about 50 million alignments per second with the affine gap model on the respective CPU. A more detailed description of this benchmark can be reviewed in the Supplementary Table S1.
3.2 Local alignment of large-scale sequences
In the second use case we aligned long genomic sequences locally as described by Liu et al. (see Table 2 for a description of the used data). We used the wavefront execution policy computing a single pairwise alignment on all 40 threads and varied the parameter for the block size. Table 3 presents the results for the block sizes that achieved the best results on each platform. We did not compare with Parasail as it cannot parallelize over a single sequence, such that we could not produce results that completed in time.
Large-scale sequences of different sizes used to evaluate the vectorized wavefront model
| ID | Accession no. | Length | Genome description |
|---|---|---|---|
| D4.4M | NC_000962.3 | 4 411 532 | Mycobacterium tuberculosis H37Rv |
| D4.6M | NC_000913.3 | 4 641 652 | Escherichia coli K12 MG1655 |
| D23M | NT_033779.4 | 23 011 544 | Drosophila melanogaster chr. 2L |
| D33M | BA_000046.3 | 32 799 110 | Pan troglodytes DNA chr. 22 |
| D42M | NC_019481.1 | 42 034 648 | Ovis aries breed Texel chr. 24 |
| D50M | NC_019478.1 | 50 073 674 | Ovis aries breed Texel chr. 21 |
| ID | Accession no. | Length | Genome description |
|---|---|---|---|
| D4.4M | NC_000962.3 | 4 411 532 | Mycobacterium tuberculosis H37Rv |
| D4.6M | NC_000913.3 | 4 641 652 | Escherichia coli K12 MG1655 |
| D23M | NT_033779.4 | 23 011 544 | Drosophila melanogaster chr. 2L |
| D33M | BA_000046.3 | 32 799 110 | Pan troglodytes DNA chr. 22 |
| D42M | NC_019481.1 | 42 034 648 | Ovis aries breed Texel chr. 24 |
| D50M | NC_019478.1 | 50 073 674 | Ovis aries breed Texel chr. 21 |
Large-scale sequences of different sizes used to evaluate the vectorized wavefront model
| ID | Accession no. | Length | Genome description |
|---|---|---|---|
| D4.4M | NC_000962.3 | 4 411 532 | Mycobacterium tuberculosis H37Rv |
| D4.6M | NC_000913.3 | 4 641 652 | Escherichia coli K12 MG1655 |
| D23M | NT_033779.4 | 23 011 544 | Drosophila melanogaster chr. 2L |
| D33M | BA_000046.3 | 32 799 110 | Pan troglodytes DNA chr. 22 |
| D42M | NC_019481.1 | 42 034 648 | Ovis aries breed Texel chr. 24 |
| D50M | NC_019478.1 | 50 073 674 | Ovis aries breed Texel chr. 21 |
| ID | Accession no. | Length | Genome description |
|---|---|---|---|
| D4.4M | NC_000962.3 | 4 411 532 | Mycobacterium tuberculosis H37Rv |
| D4.6M | NC_000913.3 | 4 641 652 | Escherichia coli K12 MG1655 |
| D23M | NT_033779.4 | 23 011 544 | Drosophila melanogaster chr. 2L |
| D33M | BA_000046.3 | 32 799 110 | Pan troglodytes DNA chr. 22 |
| D42M | NC_019481.1 | 42 034 648 | Ovis aries breed Texel chr. 24 |
| D50M | NC_019478.1 | 50 073 674 | Ovis aries breed Texel chr. 21 |
Performance evaluation of the vectorized wavefront alignment for single large-scale sequence alignments on the SKX
| SKX (t = 40; avx512) | ||||
|---|---|---|---|---|
| β | time | GCUPS | Factor | |
| D4.4M vs. D4.6M | 2000 | 137.61 | 148.81 | 816.87 |
| D23M vs. D33M | 2500 | 3155.64 | 239.18 | 1312.96 |
| D23M vs. D42M | 3000 | 3831.22 | 252.47 | 1385.95 |
| D23M vs. D50M | 3000 | 4601.75 | 250.40 | 1374.56 |
| D33M vs. D42M | 3000 | 5327.20 | 258.80 | 1420.70 |
| D33M vs. D50M | 3000 | 6384.45 | 257.25 | 1412.15 |
| D42M vs. D50M | 3000 | 8147.52 | 258.34 | 1418.16 |
| SKX (t = 40; avx512) | ||||
|---|---|---|---|---|
| β | time | GCUPS | Factor | |
| D4.4M vs. D4.6M | 2000 | 137.61 | 148.81 | 816.87 |
| D23M vs. D33M | 2500 | 3155.64 | 239.18 | 1312.96 |
| D23M vs. D42M | 3000 | 3831.22 | 252.47 | 1385.95 |
| D23M vs. D50M | 3000 | 4601.75 | 250.40 | 1374.56 |
| D33M vs. D42M | 3000 | 5327.20 | 258.80 | 1420.70 |
| D33M vs. D50M | 3000 | 6384.45 | 257.25 | 1412.15 |
| D42M vs. D50M | 3000 | 8147.52 | 258.34 | 1418.16 |
Note: The factor column is based on the GCUPS sampled for the sequential scalar algorithm on the respective platform.
Performance evaluation of the vectorized wavefront alignment for single large-scale sequence alignments on the SKX
| SKX (t = 40; avx512) | ||||
|---|---|---|---|---|
| β | time | GCUPS | Factor | |
| D4.4M vs. D4.6M | 2000 | 137.61 | 148.81 | 816.87 |
| D23M vs. D33M | 2500 | 3155.64 | 239.18 | 1312.96 |
| D23M vs. D42M | 3000 | 3831.22 | 252.47 | 1385.95 |
| D23M vs. D50M | 3000 | 4601.75 | 250.40 | 1374.56 |
| D33M vs. D42M | 3000 | 5327.20 | 258.80 | 1420.70 |
| D33M vs. D50M | 3000 | 6384.45 | 257.25 | 1412.15 |
| D42M vs. D50M | 3000 | 8147.52 | 258.34 | 1418.16 |
| SKX (t = 40; avx512) | ||||
|---|---|---|---|---|
| β | time | GCUPS | Factor | |
| D4.4M vs. D4.6M | 2000 | 137.61 | 148.81 | 816.87 |
| D23M vs. D33M | 2500 | 3155.64 | 239.18 | 1312.96 |
| D23M vs. D42M | 3000 | 3831.22 | 252.47 | 1385.95 |
| D23M vs. D50M | 3000 | 4601.75 | 250.40 | 1374.56 |
| D33M vs. D42M | 3000 | 5327.20 | 258.80 | 1420.70 |
| D33M vs. D50M | 3000 | 6384.45 | 257.25 | 1412.15 |
| D42M vs. D50M | 3000 | 8147.52 | 258.34 | 1418.16 |
Note: The factor column is based on the GCUPS sampled for the sequential scalar algorithm on the respective platform.
We can observe that for smaller sequences a smaller block size yields better results. Although D4.4M and D4.6M are over 4 Mbp long, the vectorization adds another factor to the number of blocks along the minor diagonal, which is 32 on the SKX. Thus, the block size needs to be reduced in order to produce enough work, so that all threads can execute vectorized sub-alignments. At the same time, setting the block size too small will increase the runtime as the overhead for initializing the vectorized kernel, e.g. gathering the respective buffer values from the selected tiles or transforming the sequences into vectors, becomes too large in proportion to the execution time. On the SKX we observed a block size of 3000 to perform best for long sequences giving a peak performance of GCUPS which is 1400 times faster than the sequential scalar execution. This super-linear speedup is related to caching effects and a less-optimized scalar DP kernel. In Section 3.1 of the Supplemental Material we evaluate these observations experimentally.
3.3 Aligning PacBio reads
The third use case involves the alignment of PacBio reads, which are typically very heterogeneous in their length. We used a simulated dataset using using PBSim in version 1.0.3 (Ono et al., 2013) (Table 4 lists the PBSim configuration) and a real dataset obtained from the bam file hg002_gr37_chr22.bam (ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/AshkenazimTrio/HG002_NA24385_son/PacBio_MtSinai_NIST/MtSinai_blasr_bam_GRCh37/hg002_gr37_22.bam, accessed November 14, 2017) for the genome HG002 of the Ashkenazim trio (Zook et al., 2016). In the benchmark, we realigned the PacBio reads with their corresponding reference region as if to simulate a verification step for a read mapper. The simulated data contained 66 860 sequences with the smallest sequence having a length 2341 bases and the longest 52 668 bases and in average a length of 20 011 bases. The real dataset contained 277 598 sequences with the smallest sequence having a length of 42 bases and the longest 61 989 bases and in average a length of 8238 bases.
Configuration of PBSim
| Parameter | Value |
|---|---|
| Reference | GRCH38 chr10 |
| Mode | CLR |
| qc model | Default |
| Depth | 10 |
| Length mean | 20 000 |
| Length-sd | 5000 |
| Length-min | 100 |
| Length-max | 60 000 |
| Parameter | Value |
|---|---|
| Reference | GRCH38 chr10 |
| Mode | CLR |
| qc model | Default |
| Depth | 10 |
| Length mean | 20 000 |
| Length-sd | 5000 |
| Length-min | 100 |
| Length-max | 60 000 |
Configuration of PBSim
| Parameter | Value |
|---|---|
| Reference | GRCH38 chr10 |
| Mode | CLR |
| qc model | Default |
| Depth | 10 |
| Length mean | 20 000 |
| Length-sd | 5000 |
| Length-min | 100 |
| Length-max | 60 000 |
| Parameter | Value |
|---|---|
| Reference | GRCH38 chr10 |
| Mode | CLR |
| qc model | Default |
| Depth | 10 |
| Length mean | 20 000 |
| Length-sd | 5000 |
| Length-min | 100 |
| Length-max | 60 000 |
For the simulated data we chose a block size of 2000 as trade-off for the different sequence lengths and scheduled t × l many alignments in parallel to add enough work for the vectorized wavefront model. Here, t is the number of threads and l the number of alignments packed into one vector according to the used instruction set and score width. We also compared against Parasail using the scan algorithm with fixed integer width of 32, which was the fastest configuration. The 16 bit mode could not be used, as it is not guaranteed that all alignments can be computed in the score range of 16-bit integers due to their lengths.
As can be seen in Figure 4, SeqAn outperforms Parasail in all cases on the SKX. Moreover, our generalized alignment scheduler scales very well with the number of threads reaching a peak performance of 217 GCUPS on the SKX, which is three times faster than the best result of Parasail (68 GCUPS). As a comparison, with the same instruction set SeqAn is roughly twice as fast as Parasail.
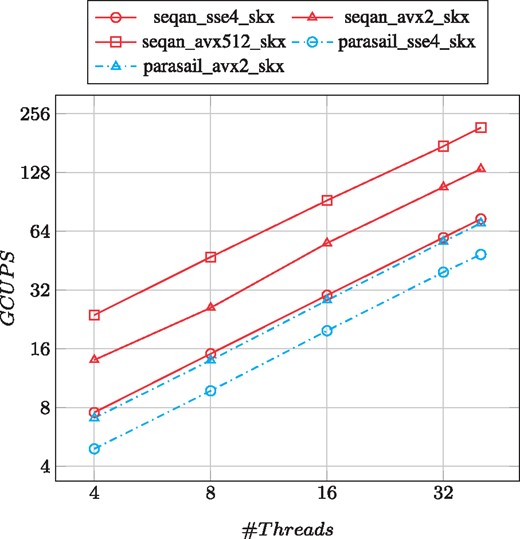
Performance and scalability comparison of aligning the PacBio-Sim data on the SKX
Table 5 shows the peak performance for the alignment of the real PacBio dataset on the SKX. We achieved the best performance with a block size of 1500. There are many more smaller sequences in the dataset, such that the optimal performance was reached with a smaller block size. However, the performance remained stable and SeqAn with SSE4 is as fast as Parasail with AVX2 and up to a factor 3 faster using AVX512.
Comparison of SeqAn’s generalized wavefront alignment with Parasail on the SKX using the PacBio-Real dataset
| SKX (t = 40) | |||
|---|---|---|---|
| Time | GCUPS | Factor | |
| seqan_sse4 | 403.76 | 65.60 | 1.6 |
| seqan_avx2 | 218.83 | 121.05 | 2.8 |
| seqan_avx512 | 137.86 | 192.14 | 4.4 |
| parasail_sse4 | 607.58 | 43.60 | 1.0 |
| parasail_avx2 | 400.35 | 66.16 | 1.5 |
| parasail_sat | 686.29 | 38.60 | 0.9 |
| SKX (t = 40) | |||
|---|---|---|---|
| Time | GCUPS | Factor | |
| seqan_sse4 | 403.76 | 65.60 | 1.6 |
| seqan_avx2 | 218.83 | 121.05 | 2.8 |
| seqan_avx512 | 137.86 | 192.14 | 4.4 |
| parasail_sse4 | 607.58 | 43.60 | 1.0 |
| parasail_avx2 | 400.35 | 66.16 | 1.5 |
| parasail_sat | 686.29 | 38.60 | 0.9 |
Note: Parasail with SSE4 was selected as the base line for the factor column.
Comparison of SeqAn’s generalized wavefront alignment with Parasail on the SKX using the PacBio-Real dataset
| SKX (t = 40) | |||
|---|---|---|---|
| Time | GCUPS | Factor | |
| seqan_sse4 | 403.76 | 65.60 | 1.6 |
| seqan_avx2 | 218.83 | 121.05 | 2.8 |
| seqan_avx512 | 137.86 | 192.14 | 4.4 |
| parasail_sse4 | 607.58 | 43.60 | 1.0 |
| parasail_avx2 | 400.35 | 66.16 | 1.5 |
| parasail_sat | 686.29 | 38.60 | 0.9 |
| SKX (t = 40) | |||
|---|---|---|---|
| Time | GCUPS | Factor | |
| seqan_sse4 | 403.76 | 65.60 | 1.6 |
| seqan_avx2 | 218.83 | 121.05 | 2.8 |
| seqan_avx512 | 137.86 | 192.14 | 4.4 |
| parasail_sse4 | 607.58 | 43.60 | 1.0 |
| parasail_avx2 | 400.35 | 66.16 | 1.5 |
| parasail_sat | 686.29 | 38.60 | 0.9 |
Note: Parasail with SSE4 was selected as the base line for the factor column.
4 Discussion
In this paper, we presented a fully generic vectorized and parallelized pairwise sequence alignment module within the SeqAn library that can be used to inherently accelerate a broad spectrum of applications in bioinformatics. Our generic design enables us to combine many variations of the core DP algorithm thus making it, to the best of our knowledge, the most comprehensive library for pairwise sequence alignments available. We provide different levels of parallelism and made them accessible through a user-friendly interface. We used this generic design to add an inter-sequence vectorization layout and combined it with an inter- and intra-sequence thread-level parallelization scheme. The tiling approach allowed us to optimize the vectorization by using 16 bit integers. This allowed us to significantly speedup the computation of many bioinformatics use cases including the alignment of PacBio reads and contigs with lengths of several mega bases. Although not specialized for any target platform, we could show that our design performs and scales overly well on general purpose CPUs as well as on high-performance manycore processor such as the Xeon® PhiTM (see Supplementary Material). In addition, we evaluated for the first time the performance of the new AVX512-BW instruction set available on the recently published Intel® Skylake processors, and showed that it effectively improves the performance in all tested use cases. In addition to exploiting the advantage of AVX512 instructions, we could further show that we substantially outperform existing implementations in any of the tested use cases on any of the used processor architectures.
Encouraged by the very good results we will steadily improve and integrate our new alignment module in several applications. For instance, we will implement a banded version of the dynamic wavefront model which can be used for example to verify PacBio reads in a PacBio aligner application more efficiently. Other applications that can be improved are our multiple sequence aligner seqan:: t-coffee (Rausch et al., 2008), which needs to progressively align hundreds of sequences, or Lambda which is high-sensitive protein aligner (Hauswedell et al., 2014).
Therefore, we will among others, improve the traceback computation for the vector-level parallelization and also develop an optimized trace method for the wavefront alignment. To compute alignments with scoring matrices more efficiently, we will also adapt the idea of a profile score as described in Rognes (2011) to our generalized inter-sequence vectorization layout. Furthermore, we will add the saturated execution mode, where in case of a score overflow or underflow the invalid alignments are recalculated with a larger integer range, which seems to work very well for protein alignments.
5 Conclusion
The need for such a library is eminently important as topical CPU architectures and HPC environments continuously increase their vectorization and multi-threading capabilities. Thus, integrating all these features into SeqAn, uniting a large set of efficient data structures and algorithms with stable and user-friendly interfaces, which are extensively tested on numerous platforms and compilers, enables application developers to exploit such high-performance systems more efficiently, paving the way for coping with the tremendous growth of data sizes, by reducing cost and time for the development of novel applications.
Acknowledgements
We gratefully thank the Konrad Zuse Institute in Berlin and in particular Thomas Steinke and Matthias Noack, for providing and assisting us in accessing their test development system running the Xeon PhiTM. Furthermore, we thank Intel® for providing us access to the newest Intel® Xeon processor and for financing the SeqAn IPCC and we thank the German Network for Bioinformatics Infrastructure (de.NBI) for financing the SeqAn infrastructure support.
Funding
This work was supported by the Intel® Parallel Compute Center at FU Berlin.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}
{kind=link}