





Abstract
FlexiDot is a cross-platform dotplot suite generating high quality self, pairwise and all-against-all visualizations. To improve dotplot suitability for comparison of consensus and error-prone sequences, FlexiDot harbors routines for strict and relaxed handling of ambiguities and substitutions. Our shading modules facilitate dotplot interpretation and motif identification by adding information on sequence annotations and sequence similarities. Combined with collage-like outputs, FlexiDot supports simultaneous visual screening of large sequence sets, enabling dotplot use for routine analyses.
FlexiDot is implemented in Python 2.7. Software and documentation are freely available at http://github.com/molbio-dresden/flexidot.
Supplementary data are available at Bioinformatics online.
1 Introduction
First described five decades ago (Gibbs and McIntyre, 1970), dotplots remain effective tools for sequence exploration, still conveying key messages in current research (Hosaka et al., 2017). Dotplots allow characterization of complex or repetitive sequences, visual detection of DNA motifs and identification of modular similarities between sequences.
Despite advances in dotplot algorithms and availability of different software tools, essential features are missing and, if available, scattered across various tools (Table 1, Supplementary Material). This prompted us to combine established functionalities (e.g. all-against-all modes) with new features for dotplot improvement (base ambiguity handling, shading, integration of annotations), while retaining usability and customizability.
Feature list of commonly used dotplot tools
| Tools | Ambiguity handling | Annotation shading | All-against-all mode | Batch analyses | Interactive GUI | Input: DNA–DNA | Input: DNA–protein | Input: protein-protein | Multiple output formats | Reverse complement | Self/pairwise collages | Similarity shading | Strict/relaxed matching | Citation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FlexiDot | + | + | + | + | – | + | – | + | + | + | + | + | +/+ | here |
| Dotmatcher | – | – | – | + | – | + | – | + | + | – | – | – | –/+ | Rice et al. (2000) |
| Dotter | – | – | + | – | + | + | + | + | – | + | – | – | +/+ | Sonnhammer and Durbin (1995) |
| Dottup | – | – | – | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| Gepard | – | + | + | – | + | + | – | + | – | + | – | – | +/– | Krumsiek et al. (2007) |
| PolyDot | – | – | + | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| YASS webserver | + | – | – | – | + | + | – | + | – | + | – | – | +/+ | Noé et al. (2005) |
| Tools | Ambiguity handling | Annotation shading | All-against-all mode | Batch analyses | Interactive GUI | Input: DNA–DNA | Input: DNA–protein | Input: protein-protein | Multiple output formats | Reverse complement | Self/pairwise collages | Similarity shading | Strict/relaxed matching | Citation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FlexiDot | + | + | + | + | – | + | – | + | + | + | + | + | +/+ | here |
| Dotmatcher | – | – | – | + | – | + | – | + | + | – | – | – | –/+ | Rice et al. (2000) |
| Dotter | – | – | + | – | + | + | + | + | – | + | – | – | +/+ | Sonnhammer and Durbin (1995) |
| Dottup | – | – | – | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| Gepard | – | + | + | – | + | + | – | + | – | + | – | – | +/– | Krumsiek et al. (2007) |
| PolyDot | – | – | + | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| YASS webserver | + | – | – | – | + | + | – | + | – | + | – | – | +/+ | Noé et al. (2005) |
GUI, Graphical user interface.
Feature list of commonly used dotplot tools
| Tools | Ambiguity handling | Annotation shading | All-against-all mode | Batch analyses | Interactive GUI | Input: DNA–DNA | Input: DNA–protein | Input: protein-protein | Multiple output formats | Reverse complement | Self/pairwise collages | Similarity shading | Strict/relaxed matching | Citation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FlexiDot | + | + | + | + | – | + | – | + | + | + | + | + | +/+ | here |
| Dotmatcher | – | – | – | + | – | + | – | + | + | – | – | – | –/+ | Rice et al. (2000) |
| Dotter | – | – | + | – | + | + | + | + | – | + | – | – | +/+ | Sonnhammer and Durbin (1995) |
| Dottup | – | – | – | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| Gepard | – | + | + | – | + | + | – | + | – | + | – | – | +/– | Krumsiek et al. (2007) |
| PolyDot | – | – | + | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| YASS webserver | + | – | – | – | + | + | – | + | – | + | – | – | +/+ | Noé et al. (2005) |
| Tools | Ambiguity handling | Annotation shading | All-against-all mode | Batch analyses | Interactive GUI | Input: DNA–DNA | Input: DNA–protein | Input: protein-protein | Multiple output formats | Reverse complement | Self/pairwise collages | Similarity shading | Strict/relaxed matching | Citation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FlexiDot | + | + | + | + | – | + | – | + | + | + | + | + | +/+ | here |
| Dotmatcher | – | – | – | + | – | + | – | + | + | – | – | – | –/+ | Rice et al. (2000) |
| Dotter | – | – | + | – | + | + | + | + | – | + | – | – | +/+ | Sonnhammer and Durbin (1995) |
| Dottup | – | – | – | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| Gepard | – | + | + | – | + | + | – | + | – | + | – | – | +/– | Krumsiek et al. (2007) |
| PolyDot | – | – | + | + | – | + | – | + | + | – | – | – | +/– | Rice et al. (2000) |
| YASS webserver | + | – | – | – | + | + | – | + | – | + | – | – | +/+ | Noé et al. (2005) |
GUI, Graphical user interface.
2 Features and implementation
FlexiDot is a multi-purpose dotplot suite for publication-ready dotplots, handling self, pairwise and all-against-all comparisons with individual and combined visualizations (Fig. 1A–C, see Supplementary Material for details). We want to highlight that (i) our mismatch and ambiguity handling enables analyses of degenerate consensus sequences and error-prone long reads (Fig. 1B), and that (ii) our sequence similarity and annotation-based shadings for self and all-against-all representations (Fig. 1A and C, respectively) convey descriptive information to facilitate sequence interpretation.
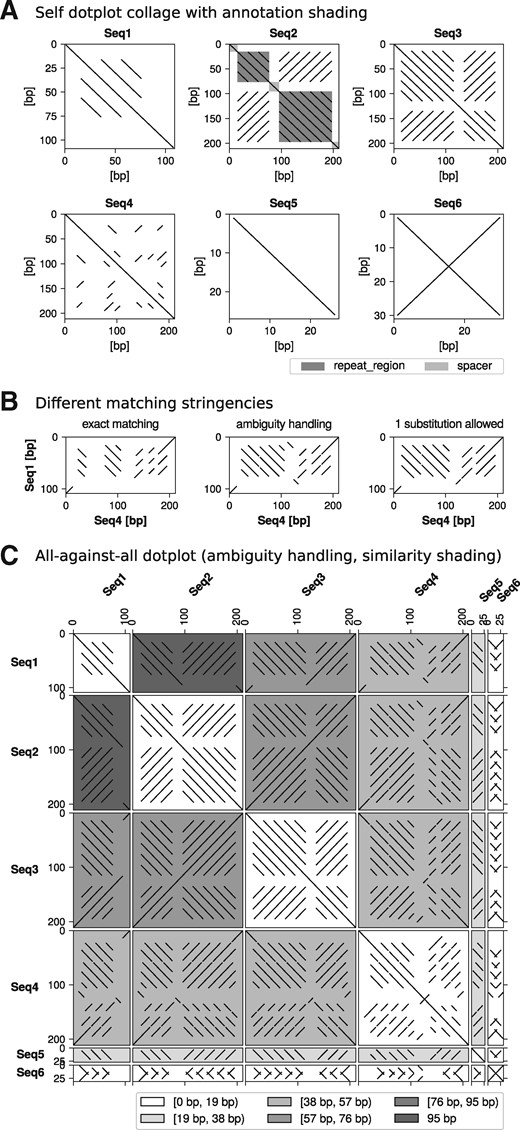
Visual sequence comparison by FlexiDot with window size 10 using six artificial test sequences. (A) Self dotplot collage. The Seq2 dotplot is shaded with custom annotations. (B) Influence of ambiguity and mismatch handling on pairwise dotplots. (C) All-against-all dotplot of the six sequences with ambiguity handling and similarity shading
The FlexiDot algorithm identifies matches, transforms them into diagonals and creates clear vector images (pdf, svg) or standard raster graphics (png). Less stringent matching is possible by addressing ambiguous residues specifically or by allowing a defined number of substitutions. A tabular output with lengths of the longest match (longest common subsequence, LCS) of all sequence pairs is provided.
FlexiDot integrates highly customizable shadings: (i) Self dotplot regions can be highlighted according to their sequence annotation provided as general feature file (Fig. 1A). (ii) All-against-all comparisons can be shaded according to the LCS length in forward, reverse or both directions (Fig. 1C). (iii) The user can provide a matrix with numerical values (e.g. identities) to guide shading. Matrix values can be displayed in the dotplot.
FlexiDot uses Python 2.7 with numpy, matplotlib, biopython, regex, colormap and colour libraries. It is operated from the command line under Windows, Linux, and Mac. Input sequences are either specified as single or multi-fasta, or automatically detected in the working directory.
3 Application
As demonstrated for a variety of use cases in the Supplementary Material, FlexiDot creates publication-ready figures for complex sequences. This facilitates:
evaluation of tandem repeat higher order structures of error-prone long reads, e.g. as seen in Sevim et al. (2016) and Symonova et al. (2017),
combined depiction of sequence structure and functional annotations,
identification of conserved motifs in related sequences,
gene or repeat comparisons using degenerated consensus sequences (Schwichtenberg et al., 2016; Weber et al., 2013),
analysis of terminal or internal inverted or direct repeats, e.g. for transposable element annotation (Hosaka et al., 2017).
Acknowledgements
We sincerely thank Michael Standke for help with the algorithm, as well as Beatrice Weber and Björn Langer for code testing and valuable feedback.
Funding
This work was supported by the German Federal Ministry of Education and Research [KMU-innovativ-18 grant 031B0224B] and the German Federal Ministry of Food and Agriculture [“Fachagentur Nachwachsende Rohstoffe e.V.” (FNR) grant 22031714].
Conflict of Interest: none declared.
References


{kind=link}