





Abstract
CRISPR-Cas9 and shRNA high-throughput sequencing screens have abundant applications for basic and translational research. Methods and tools for the analysis of these screens must properly account for sequencing error, resolve ambiguous mappings among similar sequences in the barcode library in a statistically principled manner, and be computationally efficient. Herein we present bcSeq, an open source R package that implements a fast and parallelized algorithm for mapping high-throughput sequencing reads to a barcode library while tolerating sequencing error. The algorithm uses a Trie data structure for speed and resolves ambiguous mappings by using a statistical sequencing error model based on Phred scores for each read.
The package source code and an accompanying tutorial are available at http://bioconductor.org/packages/bcSeq/.
Supplementary data are available at Bioinformatics online.
1 Introduction
High-throughput sequencing of screening assays using short hairpin RNA interference (shRNA) libraries or single-guide RNA CRISPR/Cas9 knockout (sgRNA) libraries is a powerful method for forward genetic screens. These screens have been used to study gene function in multiple diseases, including many cancer types. In the study of cancer, such screening approaches are very useful to study clonal dynamics of therapy resistance and sensitivity for targeted therapies (Doudna and Charpentier, 2014).
A key step in the analysis of the data from shRNA and CRISPR screen assays is to map each observed read in a sequenced library back to its originating barcode in the reference library. Aside from computational considerations, one has to account for sequencing error in a statistically principled manner. One approach is to repurpose tools for mapping short reads to large genomes (e.g. bowtieLangmead et al., 2009). This approach induces large overhead due to the library indexing and determining starting position for the alignment of each read. Other tools designed for mapping short reads to short barcodes are available (e.g. Dai et al., 2014; Li et al., 2014; Mun et al., 2016; Sims et al., 2011). However, these methods have two main limitations: 1. do not explicitly model the reads error distributions; and 2. are unable to properly handle ambiguous mapping (reads mapped to multiple barcodes).
In this paper, we present bcSeq an open-source R (R Core Team, 2017) extension package for fast mapping of reads from high-throughput shRNA and sgRNA sequencing assays with multi-thread support. The package resolves ambiguous mappings on the basis of a statistical model that can be customized by the user.
2 Implementation
For each read, bcSeq models the distribution of the originating barcode given the observed sequence using the corresponding Phred scores and maps the read to a reference barcode using a Bayes’ classifier. bcSeq uses a C++ implemented Trie (Bieganski et al., 1994) data structure and the pthreads model for implementing multi-threading. It provides the option for using either Hamming or edit distance. The package accepts data in FASTA and FASTQ formats for reference sequences, FASTQ for read sequences, and also interfaces with functions and classes from core Bioconductor (Huber et al., 2015) packages. The user can opt to output a tabulation of the number of reads mapped to each barcode or to output a mapping probability matrix among all the reads and barcodes in sparse format. Technical details are provided in SI.
3 Results
We conduct a simulation study to compare the performance of bcSeq to that of a perfect match approach (see Fig. 1). We consider a reference library consisting of 500 barcodes each of length 18 and a minimum pairwise edit distance of 2. We simulate 1000 sequencing libraries each consisting of 106 reads and sample the Phred scores from a real sequenced library. We evaluate the performance across all simulated reads as well as within regions of similar average Phred scores. Performance is quantified by empirical ratio of reads mapped to the library barcodes and by empirical specificity (excluding unmapped reads). We observe that compared to the perfect match approach, bcSeq consistently maps more reads with similar specificity. Furthermore, we observe that the relative gain in reads mapped for our method improves as the quality of the reads decreases. In SI, we provide additional details for the simulation study along with a performance comparison to three published methods on the basis of an L1 norm excluding unmapped reads (see Supplementary Table S2). bcSeq meets or exceeds the performance of each method considered.
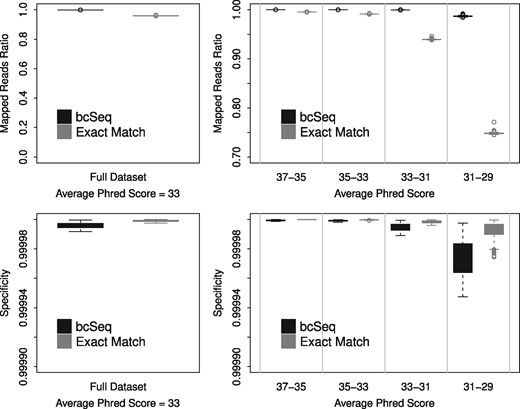
bcSeq maps more reads to barcodes for realistic data than perfect match without introducing significant mapping error. The upper left panel shows that bcSeq has a higher ratio of reads mapped to library barcodes (number of mapped reads/number of total reads) than perfect match when run on reads simulated using Phred scores sampled from real sequencing data. The upper right panel elucidates the role of sequencing error in bcSeq’s performance. Each read in the real sequencing data was binned based its average Phred score, and bcSeq and perfect match were run on reads simulated using Phred scores sampled from each of these bins. Perfect match’s mapped reads ratio degrades much more rapidly than bcSeq’s as average Phred score decreases. The lower panels compare the specificity (excluding unmapped reads) of bcSeq and perfect match. The lower left panel shows that bcSeq has a similar specificity to perfect match when run on reads simulated using Phred scores sampled from real sequencing data. The lower right panel elucidates the role of sequencing error in bcSeq’s specificity. bcSeq has a similar specificity to perfect match as the average Phred score decreases
The completion times for mapping a sequenced library consisting of 107 reads to a reference library consisting of 87 897 barcodes are 60, 31, 16, 12 and 9 min based on 1, 2, 4, 6 and 8 cores respectively on an AMD FX-8350 desktop CPU. We note these times include quality evaluation and exhaustive mapping within the mismatch threshold.
4 Discussion and conclusion
For mapping applications, as illustrated by the simulation results presented in this paper and SI, bcSeq has better performance compared to a number of other methods. The scope of its application goes beyond merely mapping barcodes, enabling rigorous statistical inference. In this context, a typical objective is to model the relationship between the relative prevalence of each barcode and a given phenotype or experimental condition. Existing methods for these analyses (e.g. MAGeCK) implicitly assume that each read is correctly mapped to its originating barcode. However, the goal of inference is to model the relationship between a phenotype and the originating, and not the mapped barcode. As discussed in SI, our approach enables rigorous inference using a measurement error model.
bcSeq is an open source R package for fast and accurate mapping of reads from high-throughput shRNA and sgRNA sequencing assays based on a default or a user-defined Phred score based probability model. The package vignette provides a worked example for customization of this model. bcSeq accommodates sequences of varying lengths and provides the option of using Hamming or edit distance, and can tolerate mismatches, internal insertions and deletions. It uses a fast Trie data structure coded in C++ and supports multi-threading. bcSeq is available through Bioconductor. The user can interface with the package using standard sequencing data formats or by using Bioconductor data structures. The code and data to reproduce the simulation and to replicate the benchmark results are described in SI.
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank two reviewers for comments and suggestions which substantially improved the paper and software package.
Funding
This research was supported in part by awards P01CA142538 (AA, KO and JL) and 5P50-CA190991-04 (KO, SF) from the National Cancer Institute.
Conflict of Interest: none declared.
References


{kind=link}