





Abstract
This study addresses several important questions related to naturally underrepresented sequences: (i) are there permutations of real genomic DNA sequences in a defined length (k-mer) and a given lineage that do not actually exist or underrepresented? (ii) If there are such sequences, what are their characteristics in terms of k-mer length and base composition? (iii) Are they related to CpG or TpA underrepresentation known for human sequences? We propose that the answers to these questions are of great significance for the study of sequence-associated regulatory mechanisms, such cytosine methylation and chromosomal structures in physiological or pathological conditions such as cancer.
We empirically defined sequences that were not included in any well-known public databases as lineage-associated underrepresented permutations (LAUPs). Then, we developed a Jellyfish-based LAUPs analysis application (JBLA) to investigate LAUPs for 24 representative species. The present discoveries include: (i) lengths for the shortest LAUPs, ranging from 10 to 14, which collectively constitute a low proportion of the genome. (ii) Common LAUPs showing higher CG content over the analysed mammalian genome and possessing distinct CG*CG motifs. (iii) Neither CpG-containing LAUPs nor CpG island sequences are randomly structured and distributed over the genomes; some LAUPs and most CpG-containing sequences exhibit an opposite trend within the same k and n variants. In addition, we demonstrate that the JBLA algorithm is more efficient than the original Jellyfish for computing LAUPs.
We developed a Jellyfish-based LAUP analysis (JBLA) application by integrating Jellyfish (Marçais and Kingsford, 2011), MEME (Bailey, et al., 2009) and the NCBI genome database (Pruitt, et al., 2007) applications, which are listed as Supplementary Material.
Supplementary data are available at Bioinformatics online.
1 Introduction
For a given length of DNA comprising the four natural nucleotides A, T, G and C, all possible sequences of the same length but not in the same nucleotide order are referred to as sequence permutations (Bujnicki, 2002; Gill and Machattie, 1976; Jeltsch, 1999). Due to the existence of covalent nucleotide modifications, such as the methylation and hydroxymethylation of cytosine, these permutations are limited only to sequences composed of the four primary nucleotides (Koskinen, 2012). However, when we insist on a true distribution of all possible permutations in the current best collections comprising gigabase-magnitude genomic sequences that include the most dominant public DNA databases, such as GenBank, EMBL and DDBJ (Ouellette, 1998; Stoesser et al., 1999; Tateno et al., 2002), a subset of DNA sequences appears to be underrepresented; they are neither sequence defects nor errors since their abundance is above both thresholds. Such underrepresentation is expected to have broad indications, including sequence-specific regulatory motifs and chromosomal DNA or RNA structural landmarks, such as telomeric repeats of GGATTT. The underrepresented sequences have further demonstrated to be lineage-associated when examined over broad taxa.
Hampikian and Anderson (Hampikian and Andersen, 2007) initially defined these aforementioned sequences as nullomers. Subsequently, many scientists (Acquisti et al., 2007; Herold et al., 2008; Vergni and Santoni, 2016) continued Hampikian and Anderson’s study and focused on the origin of nullomers. Here, we empirically define such sequences that have never existed in any well-known public databases as lineage-associated underrepresented permutations (LAUPs). Notably, the widely used public databases for this study include GenBank, EMBL and DDBJ (Ouellette, 1998; Stoesser et al., 1999; Tateno et al., 2002).
This study defines these aforementioned sequences as LAUPs, but not nullomers (Hampikian and Andersen, 2007), because of four considerations. First, nullomers or LAUPs are similarly defined if both sequences are limited to a particular lineage, but the former emphasizes the absence of a sequence and the latter emphasizes the characteristics of a sequence. The nullomers from mammals or even vertebrates are similar and may be classified as well as traced to some ancestral groups; however, if genomes of other lineages are analysed in a similar manner, then the nullomers are different, such as between vertebrates and arthropods. Second, when sequence variations in a population background are categorized, an allelic definition must be introduced. In such a context, a nullomer and its variants may be classified into major or minor alleles; if a nullomer becomes a minor allele and one of its variants becomes the major allele, then naming the nullomer is inappropriate. Third, since sequencing platforms consistently have error rates, nullomer sequences become empirically defined because a fraction of the nullomers may result from sequencing errors. Fourth, since the goals of this study are not limited to only nullomer sequences, we may have to define closely related sequences based on structural and conformational similarities. Thus, it is rather precise to use an underrepresented permutation notation to define these sequences.
The compositional dynamics, or degree of sequence variability, of DNA sequences, particularly human DNA sequences, are slightly biased in TpA (Yomo and Ohno, 1989) and CpG (Gardiner-Garden and Frommer, 1987). The best known CpG enrichments are the so-called CpG islands (CGI) sequences that are not only partitioned among half of the housekeeping genes (Byun et al., 1999; Daniel Eller, 2007; Farré, 2007; Gardiner-Garden and Frommer, 1987; Lawson and Zhang, 2008; Yang et al., 2015) but also regulate ∼70% of the mammalian genome (Han et al., 2008). Most CGI sequences are associated with the 5′ ends of house-keeping genes (Tykocinski and Max, 1984). CGI sequences are defined as those with the observe/expectation values >0.6 and whose GC content is >50% (Gardiner-Garden and Frommer, 1987; Takai and Jones, 2002).
Recent studies have indicated (Pan et al., 2017; Pongor et al., 2017; Yu et al., 2013) that CpG sequence underrepresentation is largely due to the instability of cytosine methylation (Thellin et al., 1999). Since both LAUPs and CGI sequences are underrepresented, we question if a connection exists between LAUPs and CGI sequences.
To investigate the connection between LAUPs and CGI sequences, we developed a Jellyfish-based LAUPs analysis application (JBLA) for efficient LAUPs computation by integrating several relevant software tools (Bailey et al., 2009; Rozenberg et al., 2008). Thus, JBLA not only investigates the CG content and motif of LAUPs as a bridge for studying CGI sequences but also for examining CG permutations in both LAUPs and CGI sequences.
This study generated the following interesting findings: (i) length ranges of the shortest LAUPs from 24 representative species, (ii) higher CG content and enriched CG*CG motifs among common LAUPs found in the mammalian genome and (iii) relatedness between GC-rich LAUPs and CGI sequences. In general, this study focused on the motifs and sequence composition patterns between LAUPs and CGI sequences since both LAUPs and CGI sequences are CG-enriched and underrepresented.
2 Materials and methods
2.1 Data source
The sequence data for this study included those of 24 representative species (Supplementary Table S1) from NCBI sites (Pruitt et al., 2007) and were the latest versions of the GenBank GBFF (GenBank Flat File) format (Ouellette, 1998). Supplementary Table S1 lists the names, data sizes and genome data versions for all 24 species. We divided the 24 species into 5 lineages: (i) Bacteria, (ii) Plants, (iii) Human, (iv) Primates (other than Human) and (v) Mammals (excluding Primates). We used a Perl script to remove unnecessary annotations and extract the genome sequence data, and we used the sequences and their reverse-complement sequences (Segerstéen et al., 1986) in this analysis. Notably, this study focused on lineage-associated species and analysed common LAUPs for the same lineage to avoid the impact of different genome sizes because it is difficult to normalize the genome sizes for different lineages.
2.2 LAUP analysis algorithm
The LAUPs analysis algorithm is shown in Figure 1, which comprises LAUPs computing (Supplementary Fig. S1A) and data analysis components (Supplementary Fig. S1B and C). We detail the components and define several important terms for the analysis in Table 1.
Terms for sequence analysis
| Definition | Description |
|---|---|
| LAUPs | The sequences that never exist in major public databases with respect to a given lineage |
| Sim_set | Comprises all possible 4K k-mers. |
| Kwg_set | Collects the k-mers from the existing public databases of the species |
| LAUPs_set | The complement of Kwg_set with respect to Sim_set |
| The shortest LAUPs’ average proportion (Fig. 2) | The number of the shortest LAUPs over that of all permutations |
| CpG-containing sequence | The sequence comprising CG*, where * indicates any nucleotides |
| CommonLAUPs | LAUPs intersection set for 21 mammalian species |
| Observe (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k. Here, n is the number of CpGs in a k-mer, and k is the length of a k-mer |
| Expect (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k in the random condition (Supplementary Method 1.4) |
| OE_Ratio (k, n) | The ratio between Observe (k, n) and Expect (k, n), detailed in Equation (4) |
| Positive state (k, n) | If OE_Ratio (k, n) is >1 |
| Negative state (k, n) | If OE_Ratio (k, n) is ≤l |
| Definition | Description |
|---|---|
| LAUPs | The sequences that never exist in major public databases with respect to a given lineage |
| Sim_set | Comprises all possible 4K k-mers. |
| Kwg_set | Collects the k-mers from the existing public databases of the species |
| LAUPs_set | The complement of Kwg_set with respect to Sim_set |
| The shortest LAUPs’ average proportion (Fig. 2) | The number of the shortest LAUPs over that of all permutations |
| CpG-containing sequence | The sequence comprising CG*, where * indicates any nucleotides |
| CommonLAUPs | LAUPs intersection set for 21 mammalian species |
| Observe (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k. Here, n is the number of CpGs in a k-mer, and k is the length of a k-mer |
| Expect (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k in the random condition (Supplementary Method 1.4) |
| OE_Ratio (k, n) | The ratio between Observe (k, n) and Expect (k, n), detailed in Equation (4) |
| Positive state (k, n) | If OE_Ratio (k, n) is >1 |
| Negative state (k, n) | If OE_Ratio (k, n) is ≤l |
Terms for sequence analysis
| Definition | Description |
|---|---|
| LAUPs | The sequences that never exist in major public databases with respect to a given lineage |
| Sim_set | Comprises all possible 4K k-mers. |
| Kwg_set | Collects the k-mers from the existing public databases of the species |
| LAUPs_set | The complement of Kwg_set with respect to Sim_set |
| The shortest LAUPs’ average proportion (Fig. 2) | The number of the shortest LAUPs over that of all permutations |
| CpG-containing sequence | The sequence comprising CG*, where * indicates any nucleotides |
| CommonLAUPs | LAUPs intersection set for 21 mammalian species |
| Observe (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k. Here, n is the number of CpGs in a k-mer, and k is the length of a k-mer |
| Expect (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k in the random condition (Supplementary Method 1.4) |
| OE_Ratio (k, n) | The ratio between Observe (k, n) and Expect (k, n), detailed in Equation (4) |
| Positive state (k, n) | If OE_Ratio (k, n) is >1 |
| Negative state (k, n) | If OE_Ratio (k, n) is ≤l |
| Definition | Description |
|---|---|
| LAUPs | The sequences that never exist in major public databases with respect to a given lineage |
| Sim_set | Comprises all possible 4K k-mers. |
| Kwg_set | Collects the k-mers from the existing public databases of the species |
| LAUPs_set | The complement of Kwg_set with respect to Sim_set |
| The shortest LAUPs’ average proportion (Fig. 2) | The number of the shortest LAUPs over that of all permutations |
| CpG-containing sequence | The sequence comprising CG*, where * indicates any nucleotides |
| CommonLAUPs | LAUPs intersection set for 21 mammalian species |
| Observe (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k. Here, n is the number of CpGs in a k-mer, and k is the length of a k-mer |
| Expect (k, n) | The number of k-mer(s) that consist of an n number of CpGs for each k in the random condition (Supplementary Method 1.4) |
| OE_Ratio (k, n) | The ratio between Observe (k, n) and Expect (k, n), detailed in Equation (4) |
| Positive state (k, n) | If OE_Ratio (k, n) is >1 |
| Negative state (k, n) | If OE_Ratio (k, n) is ≤l |
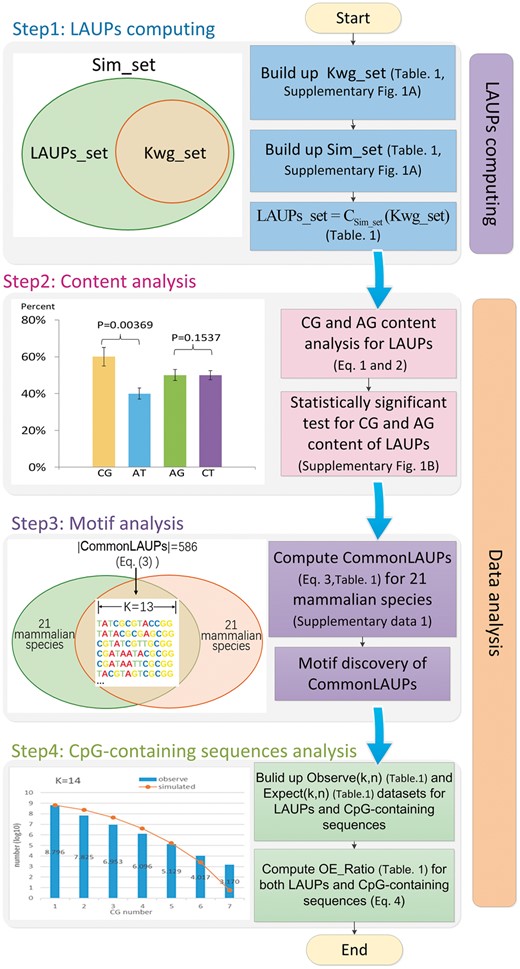
Workflow of the LAUPs analysis algorithm. Here, the terms of Kwg_set, Sim_set and LAUPs_set are detailed in Table 1 (Color version of this figure is available at Bioinformatics online.)
2.2.1 LAUPs computing component
This component was used to obtain a set of LAUPs for each species, such as Homo sapiens and Pan troglodytes (Pruitt et al., 2007). The related algorithm is detailed in Supplementary Method 1.1.
2.2.2 Data analysis component
This component consists of content analysis process, motif analysis and CpG-containing sequences analysis scenarios.
(1) Content analysis: we computed the CG and AT content proportion as well as the purine (AG) and pyrimidine (TC) content proportion of LAUPs. We employed a well-developed statistical testing process (Zhang et al., 2017a, b) to validate the content statistical significance between CG and AT as well as the difference between purine (AG) and pyrimidine (TC) for LAUPs. The related algorithm is detailed in Supplementary Method 1.2.
(2) Motif analysis process: sequence motifs are short and recurring patterns in DNA sequences with potential biological function (D’Haeseleer, 2006). The motif search procedure is called motif discovery (Bailey and Elkan, 1994). This study employed a motif search tool, MEME (Bailey et al., 2009), to search for frequently occurring or common LAUP patterns of various species.
(3) CpG-containing sequences analysis: this analysis investigated the CG frequency between commonLAUPs and CpG-containing human sequences. The related algorithm is detailed in Supplementary Methods 1.3 and 1.4.
3 Results
3.1 The shortest LAUPs for the representative species
To define sequence permutations of different lengths or k-mers in this study, we first built a dataset that contained all possible sequence permutations, and subsequently searched for sequence permutations at a given k-mer length of a genomic sequence to investigate their representations, CG contents, motifs and proportions among the permutations. Although we conducted this analysis for most species, this study focused on mammalian genome sequences, which are further partitioned into three categories: humans, primates (non-human primates) and mammals (non-primate mammals); the genomes of plants and bacteria were used as controls (Supplementary Table S1).
We show that the length of the shortest LAUPs lies between 10 and 14 for whole genomes (WG) (Fig. 2A), and the shortest LAUPs average proportion rapidly and non-linearly increases with increasing length of the sequences (Fig. 2B and Supplementary Fig. S2A).
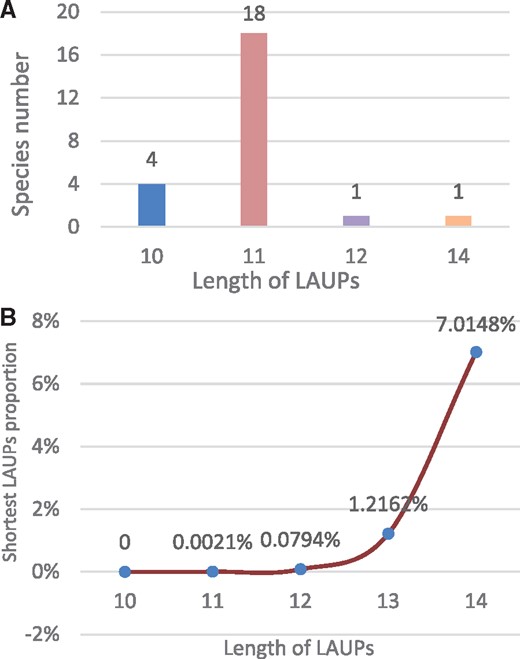
The shortest LAUPs for the representative species. (A) The number of species for the shortest LAUPs. (B) The average proportion of the shortest LAUPs (denoted in Table 1) (Color version of this figure is available at Bioinformatics online.)
3.2 GC and AG content analysis for LAUPs
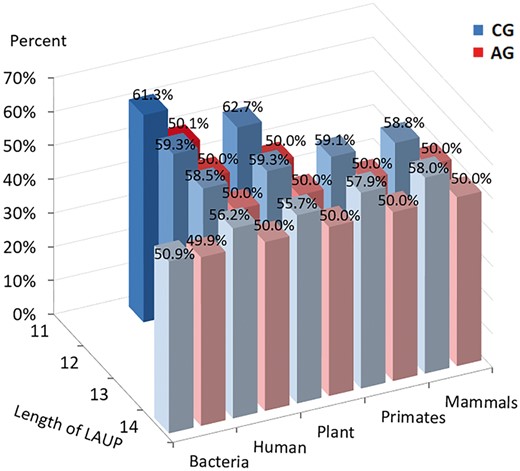
AG (purine) and GC contents of some common LAUPs (Color version of this figure is available at Bioinformatics online.)
3.3 Motif analysis for LAUPs
The length of the shortest LAUPs for the mammalian lineage is 13, and CommonLAUPs consists of 586 LAUPs (left figure of Step 3 in Fig. 1). We also investigated putative motifs among the common LAUPs (Bailey et al., 2009) by Motif Discovery logos (Schneider and Stephens, 1990). Figure 4 reveals a striking common characteristic: the CpG dinucleotide sequence appeared biased towards a series of CG*CG sequences.
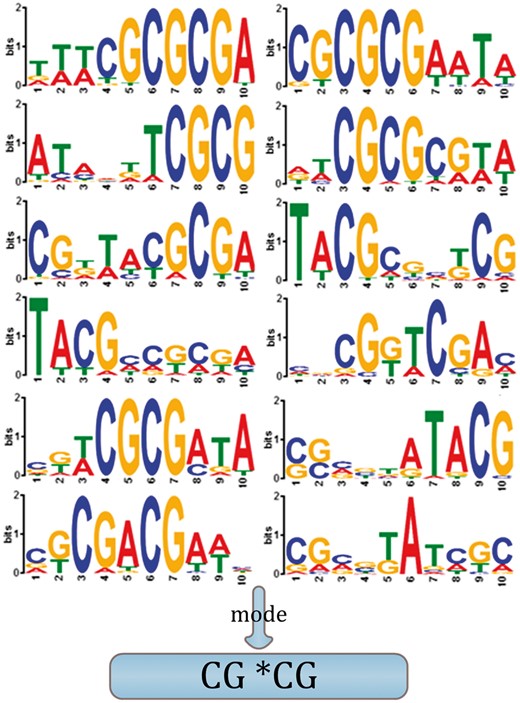
Motif discovery logos for 21 typical mammalian species. The horizontal and vertical axes represent the position of the motif and its ‘bits per position’ (D’Haeseleer, 2006), respectively (Color version of this figure is available at Bioinformatics online.)
3.4 CpG-containing sequences analysis
The LAUPs among mammals had a higher GC content, consistent with that of CGI sequences (Gardiner-Garden and Frommer, 1987; Takai and Jones, 2002). Further examination revealed a pattern of CpG*CpG in most high-GC LAUPs (Fig. 3). Since CGI sequences may play regulatory roles (Yu et al., 2013; Zhu et al., 2008), we examined the relatedness of the two GC-rich sequence groups, LAUPs versus CGI, in terms of structural and conformational features. Here, we investigated the CGI definition (Gardiner-Garden and Frommer, 1987; Takai and Jones, 2002) and explored permutation patterns for the CGI sequences, considering only the CpG density but not the CpG permutation. However, CGI sequences are typically too long and too abundant to directly examine their permutation patterns; therefore, we selected short LAUPs as a bridge for the present comparative analysis.
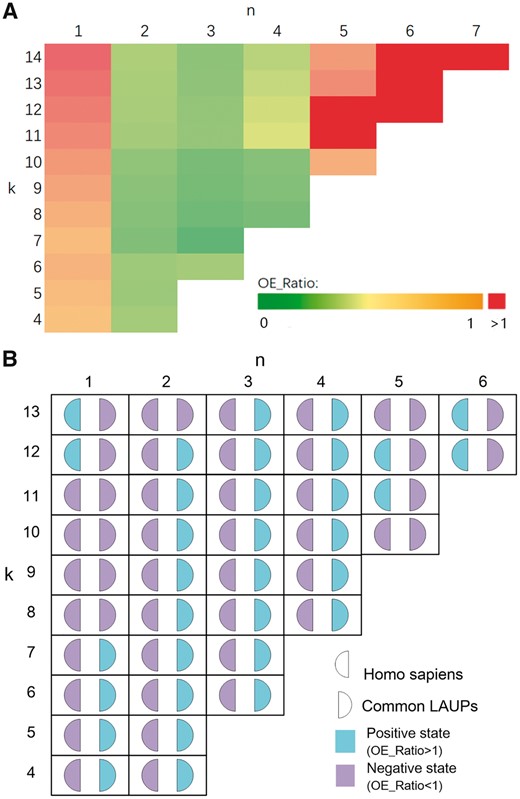
OE_Ratio. (A) An OE_Ratio heat map for CpG-containing human sequences; (B) comparison between human CpG-containing sequences and common LAUPs (Color version of this figure is available at Bioinformatics online.)
3.5 Algorithm performance comparison
In this study, we developed a JBLA to analyse LAUPs sequences with two major components (Fig. 1): LAUPs computation and data analysis. To compute LAUPs more efficiently, JBLA has improved Jellyfish (Marçais and Kingsford, 2011) in the following three aspects.
We compared the LAUPs computing efficiency between JBLA and Jellyfish by selecting three species (Oryza sativa, Microcebus murinus and Miniopterus natalensis) as the test datasets, employing both JBLA and Jellyfish to compute LAUPs 10 times when k is from 9 to 11, and by using statistical tests (Fig. 6) to demonstrate statistically significant differences between JBLA and Jellyfish.
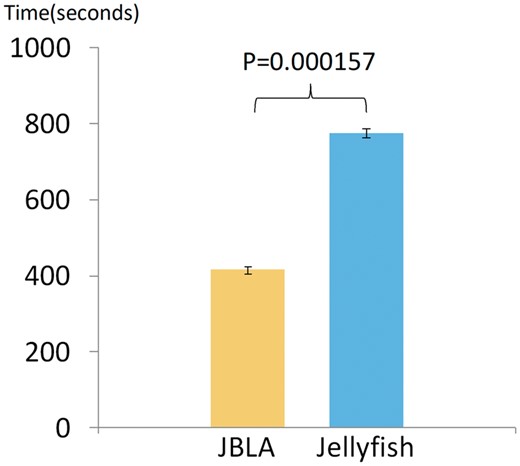
The computing efficiency between JBLA and Jellyfish (Color version of this figure is available at Bioinformatics online.)
4 Discussion
Since CGI and LAUP sequences are both underrepresented among mammalian genomes, we analysed short LAUPs and explored their CpG-containing sequence permutations by using JBLA between the two GC-rich sequences.
We first revealed the length of the shortest LAUPs ranges from 10 to 14 (Fig. 2A andSupplementary Fig. S2). There are two factors considered relevant to this size range. One factor is the uniqueness or complexity of genome sequences; as far as mammalian genomes are concerned, k = 15 is the lower bound of such complexity, ∼3×109 bp (415). The other factor is the physical force for base pairing. The force of the shortest LAUPs should be between 10*14 = 140 pN and 14*20 = 280 pN, consistent with Zhang et al. (2015) that the base-pairing strength of single dG/dC and single dA/dT are 20.0 ± 0.2 pN and 14.0 ± 0.3 pN, respectively. Similarly, Essevaz-Roulet et al. (Essevaz-Roulet et al., 1997) and Clausen-Schaumann et al. (Clausen-Schaumann, et al., 2000) indicate that the maximum rupture forces of DNA duplexes are 300 pN, which are slightly greater than the low limit of the shortest mechanical force of LAUPs. Therefore, we consider that the length of the shortest LAUPs is mechanistically meaningful and reasonable, which could be used as a threshold for further research. Moreover, Figure 2 shows that the lengths of the shortest LAUPs for most species are ∼11 for most species. We consider that this interesting length range should be related to the length of a complete double helix, which is ∼10 (Chen et al., 2008; Worning et al., 2000).
We also consider that shortest LAUPs may start as unique sequences or become distributed over only certain limited sequence contexts because proportionally, some shortest LAUPs must be generally rare (Fig. 2B andSupplementary Fig. S2C), and these rare and unique sequences should be related to sequence-associated regulation mechanisms under physiological or pathological conditions.
Sequence characteristics are partitioned into two simple compositions, the GC and purine (A + G or simply AG) contents. We not only observe obvious higher GC content in the shortest LAUPs (Fig. 3; Supplementary Tables S2 and S3; Supplementary Fig. S1B) but also the higher GC content highlights CpG sequences rather GpC (Schweitzer and Kool, 1995). We further examined the sequence patterns among the LAUPs discovers a CG*CG mode from the common LAUPs of mammalian genomes (Fig. 4). Thus, consecutive CpG dinucleotides immediately prompted us to examine CGI sequences.
Since most CGI sequences are too long to directly compare with LAUPs for permutation patterns, we use short LAUPs and their CpG-containing sequences as a bridge to investigate those of CGI sequences (left figure of Step 3 in Fig. 1). Here, we use expected CpG-containing sequences to represent the sum of CGI sequences and non-CGI CpG-containing sequences. Obviously, only a true and de novo CGI sequence analysis provided an answer for the relatedness between CGI and LAUP sequences.
The results of this study are multi-fold. First, since both observed and expected CpG-containing sequences have frequency information attached, we determined that these sequences are not random (Fig. 5A) and their CpG*CpG modes and motifs are related to the two parameters (k and n). Second, when LAUPs have a positive state, most CpG-containing sequences show a negative state (Fig. 5B). Thus, their CpG*CpG motifs and modes appear to mirror each other, suggesting the existence of strong sequence or sequence motif selections, preserving or excluding some of the permutations and patterns within, which may serve as unique motifs for both trans and cis regulations, which are typically involved in protein-based recognition and/or chromosomal structural conformation, respectively. More detailed analysis is certainly expected, focusing on both CGI and LAUPs directly to search for meaningful sequence motifs and patterns, followed by experimental evaluation. A final notion is attributable to JBLA, which is much more efficient than Jellyfish for LAUPs computing (Fig. 6) and secures future analysis based on larger datasets.
Although this study explores several interesting findings based on the characteristics of LAUPs, further investigation is still needed. For example, we examined LAUPs and their variants in the context of pan-cancer genomes for regulatory sequence changes. In addition, genome-wide DNA sequences are abundant, but the cost to analyse these sequences are computing-intensive and time-consuming. Also, we do not have a database that hosts both species- and lineage-associated LAUPs from other sequenced genomes, which can offer a platform for other data analysis activities, such as data distribution and visualization. Therefore, our further study will employ high performance computing (Jiang et al., 2015; Jiang et al., 2011) and related data mining algorithms(Gao et al., 2017; Jiang et al., 2015; Jiang et al., 2011; Peng et al., 2014; Zhang et al., 2017a, b; Zhang et al., 2016) to speed up the Jellyfish software and build up a LAUPs warehouse in the distant future.
Acknowledgements
This work was supported by the General Program from National Natural Science Foundation of China, Chongqing excellent youth award and the Chinese Recruitment Program of Global Youth Experts as well as by the Fundamental Research Funding of the Chinese Central Universities.
Funding
This work was supported by National Natural Science Foundation of China [61372138], the National Science and Technology Major Project [2018ZX10201002], Chongqing Research Program of Basic Research and Frontier Technology [cstc2015jcyjA40026, No. cstc2016jcyjA0568] and Chinese Chongqing Distinguish Youth Funding [cstc2014jcyjjq40003].
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that Le Zhang and Ming Xiao authors contributed equally.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}