





Abstract
Classification of prokaryotic species is usually based on sequence similarity thresholds, which are easy to apply but lack a biologically-relevant foundation. Here, we present ConSpeciFix, a program that classifies prokaryotes into species using criteria set forth by the Biological Species Concept, thereby unifying species definition in all domains of life.
ConSpeciFix’s webserver is freely available at www.conspecifix.com. The local version of the program can be freely downloaded from https://github.com/Bobay-Ochman/ConSpeciFix. ConSpeciFix is written in Python 2.7 and requires the following dependencies: Usearch, MCL, MAFFT and RAxML.
1 Introduction
The most common method for defining species is the Biological Species Concept (BSC) (Mayr, 1942), which assigns individuals as members of the same species by their ability to reproduce with one another. But because the BSC relies on sexual reproduction (i.e. gene flow, homologous recombination), it is not thought to be applicable to prokaryotes, i.e. bacteria and archaea, which reproduce asexually. However, gene exchange is now known to be prevalent among prokaryotes (Shapiro and Polz, 2015; Vos and Didelot, 2009) opening the possibility that prokaryotes can be classified into species according to the BSC (Bobay and Ochman, 2017). To this end, we developed a unique tool, ConSpeciFix, that uses genomic information to classify organisms into species based on gene flow. In this context, ‘gene flow’ refers to homologous recombination events among genes in the core genome, i.e. single-copy orthologs that are shared by at least 85% of the strains considered. This contrasts recent approaches that assess species boundaries based on non-homologous gene transfers (Moldovan and Gelfand, 2018). The ConSpeciFix pipeline is available on GitHub and also implemented on a webserver, allowing users to directly upload and classify microbial genomes against a pre-processed database.
2 Materials and methods
Our procedure consists of testing for gene flow between candidate genomes and the genomes in our database, which have already been assigned to species. Gene flow is estimated by the inference of homoplasic alleles (h) relative to non-homoplasic alleles (m). Homoplasies are alleles that are not inherited vertically from a single ancestor and likely result from gene flow. We infer homoplasies with a distance-based approach: For each set of genomes, we first compute the pairwise distances D on the concatenate of the core genome using a maximum likelihood method (Stamatakis, 2006). In the simple scenario in which only two variants are present at a site, we classify the more frequent variant as the major allele (N0) and the least frequent as the minor allele (N1). We then compare the core genome distances between strains harboring alleles N1 and N0. The minor allele is considered homoplasic if max(DN1N1) > min(DN0N1), where max(DN1N1) represents the maximal core genome distance between strains harboring the minor allele and min(DN0N1) represents the minimal core genome distance between pairs of strains harboring the minor allele and the major allele. When more than three or four alleles are present at a site, multiple minor alleles are defined, and each is compared to the major allele (without including the other minor alleles in the comparison). Because homoplasies can also be introduced by independent convergent mutations, we include a feature to display the proportion normally attributable to this process based on simulations computed across 93 prokaryotic species.
3 Implementation
ConSpeciFix is hosted online at www.conspecifix.com. This server accommodates individual user-uploaded genomes, which can be evaluated for gene flow against a pre-processed database of curated species. When analyzing multiple genomes (e.g. in endeavors to define new species), a variant of ConSpeciFix and installation instructions can be downloaded from GitHub.
Database building. To build the pre-processed database, we re-classified all complete genomes of prokaryotes available on the NCBI RefSeq repository as of October 2017. We downloaded the genomes for each named species represented by 15 or more genomes. (Analyses based on fewer than 15 genomes generally do not yield robust estimates of gene flow.) A core genome was defined for each species, and gene flow was estimated with a distance-based approach, as in Bobay and Ochman (2017). Within each named species, genomes with substantially lower h/m ratios were removed from the dataset, and all remaining genomes were considered members of the same BSC-defined species. The redefined dataset of species currently contains 11 127 genomes and 236 BSC-defined species.
Testing candidate genomes. Our webserver allows users to test candidate genomes for gene flow against any of the designated BSC-defined species in the database. After selecting one of the 236 species and uploading the query genome (which can be complete or incomplete), ConSpeciFix identifies the core genes shared between the candidate genome and the selected species using Usearch Global v6.1 (Edgar, 2010). This core genome is used to re-estimate the distance matrix with RAxML v8.0 (Stamatakis, 2006), from which homoplasic and non-homoplasic alleles are inferred. Ratios of homoplasic and non-homoplasic alleles (h/m) are computed for randomly sampled subsets of genomes; and from this step, graphs and statistics comparing h/m ratios between sets of genomes that include and exclude the candidate genome are generated. Finally, ConSpeciFix estimates whether the candidate genome should be considered as part of the selected species based on an exclusion criterion by testing whether the inclusion of the candidate genome leads to a substantial drop in gene flow. The stringency of our original exclusion criterion (Bobay and Ochman, 2017) sometimes resulted in a failure to identify sexually-isolated strains in cases where the h/m graph did not quickly reach a plateau. To address this issue, the exclusion criterion now relies on the Chi2 outlier method (Dixon, 1950) implemented in the R package ‘outliers’, which allows detection of sexually-isolated strains when h/m graphs do not quickly reach a plateau. Additional metrics are returned to the user, such as the number of orthologous genes shared between the candidate genome and the number of core genes shared with the tested species. Note that our database includes numerous clonal species, which are not expected to engage in gene flow even when strains have very similar genomes. Clonal species can be identified based on their consistently low h/m ratios (typically below 0.2), as expected under evolution strictly driven by mutations (red lines in graphs, Fig. 1). For such species, we recommend that users resort to the average nucleotide identity (ANI) metric that are returned in the result statistics, a metric that is commonly applied to define prokaryotic species based on genomic data (Konstantinidis and Tiedje, 2005).
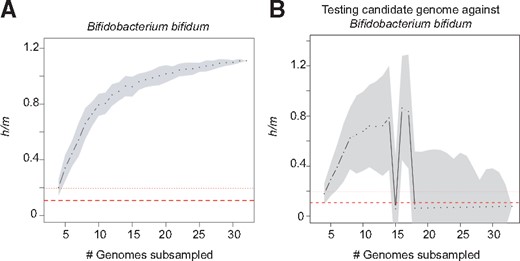
ConSpeciFix output graphs with and without the tested candidate genome. Graphs show h/m ratios estimated across different subsets of B.bifidum genomes (A) and across different subsets of B.bifidum genomes that include the B.longum 2-2B genome (B). For each number of subsampled genomes (x-axis), black dots represent the median h/m ratio estimated for 100 samplings, with grey areas indicating the standard deviation. Dashed and dotted lines indicate average and maximal h/m ratios, respectively, expected when homoplasies are generated solely by convergent mutations, as estimated for 93 species (Bobay and Ochman, 2017)
Exploratory phase. Users can forego the initial step of species selection and compare candidate genomes against an extended database (which includes our redefined dataset of 236 species plus a representative genome for each additional species present in RefSeq, yielding a total of 7105 species) to determine which species might be most suitable for evaluation. Using the ‘exploratory phase’ option, ConSpeciFix detects the most closely related species in the extended database based on a set of 45 protein coding genes that are shared across prokaryotes (Raymann, et al., 2015) by computing the number of orthologous genes shared with each species and their average nucleotide identity.
4 Application
To illustrate the ConSpeciFix process, we tested whether Bifidobacterium longum strain 2-2B engages in gene flow with strains of Bifidobacterium bifidum. The analysis returns two main graphs, one (Fig. 1A) showing the estimated rate of gene flow (with h/m ratios) across randomly sampled subsets of B.bifidum genomes, and a second (Fig. 1B) representing h/m ratios computed across subsets of B.bifidum genomes but including the tested B.longum genome. This example shows that inclusion of the B.longum 2-2B genome induces a sharp drop in h/m ratios, indicating that this strain does not engage in gene flow with B.bifidum.
Acknowledgements
We thank Kim Hammond for assistance with figures and two anonymous reviewers.
Funding
This work was supported by NIH grant R35GM118038 to HO.
Conflict of Interest: none declared.
References


{kind=link}