





Abstract
The advent of next-generation sequencing has boosted the analysis of bacterial genome evolution. Insertion sequence (IS) elements play a key role in prokaryotic genome organization and evolution, but their repetitions in genomes complicate their detection from short-read data.
PanISa is a software pipeline that identifies IS insertions ab initio in bacterial genomes from short-read data. It is a highly sensitive and precise tool based on the detection of read-mapping patterns at the insertion site. PanISa performs better than existing IS detection systems as it is based on a database-free approach. We applied it to a high-risk clone lineage of the pathogenic species Pseudomonas aeruginosa, and report 43 insertions of five different ISs (among which three are new) and a burst of ISPa1635 in a hypermutator isolate.
PanISa is implemented in Python and released as an open source software (GPL3) at https://github.com/bvalot/panISa.
Supplementary data are available at Bioinformatics online.
1 Introduction
Insertion Sequences (ISs) are the smallest transposable elements (TEs) and are widespread throughout all domains of life (Siguier et al., 2014). The common IS structure consists of (i) one or two transposase-encoding genes, (ii) two terminal inverted repeats (IRs) and (iii) two direct repeated sequences (DRs; Siguier et al., 2015). ISs are independently mobilizable. The classification of ISs mostly relies on the amino-acid similarity of their transposases (Mahillon and Chandler, 1998). In 2017, the ISfinder database reported 4000 ISs belonging to 29 families (Mahillon and Chandler, 1998; Siguier et al., 2015).
IS insertion can result in gene disruption and modulation of the expression of neighboring genes by disruption of the promoter or creation of an alternative promoter (Vandecraen et al., 2017). IS transposition enables the host to adapt to new environmental challenges and colonize new niches (Vandecraen et al., 2017). Most examples of IS transposition are linked to antibiotic resistance because related phenotypes are easy to detect. For example, the insertion of IS1 or IS10 upstream of the efflux pump acrEF increases the resistance of Salmonella enterica to fluoroquinolones (Olliver et al., 2005). Similarly, inactivation of the gene oprD by an IS induced the resistance to imipenem in clinical strains of Pseudomonas aeruginosa (Sun et al., 2016). In addition, IS transposition can affect bacterial virulence and metabolism (Siguier et al., 2014; Vandecraen et al., 2017). These mobile elements also affect the architecture of host genomes. Indeed, ISs can be recognized by the host cell recombination machinery, allowing their amplification and recombination events between individual copies, leading to deletion, inversion and duplication of portions of the host genome (Vandecraen et al., 2017).
The detection of such sequences is crucial in evolutionary studies, as IS activity is one of the most dynamic forces at play in bacterial genome modification. Next-generation sequencing of whole bacterial genomes to analyze genomic evolution and structure has become routine, with Illumina being the most commonly used technology. NGS pipeline analysis can easily detect SNPs or small insertions/deletions by aligning reads against reference genomes. In contrast, IS analysis is more complicated due to the fact that this element is repeated and that the read length (<300 bp) is usually shorter than that of ISs. Several bioinformatics tools have been developed to overcome this problem (Ewing, 2015). They are based on two different approaches: (i) Structural variant tools (i.e. DD_DETECTION or TIDDIT) search break junctions in the genome to detect potential insertions or deletions (Eisfeldt et al., 2017; Kroon et al., 2016). But the identification of the sequence inserted needs further efforts; (ii) Other tools like ISMapper or RetroSeq identify the insertion of TEs based on a query TE database as input, preventing the detection of unknown elements (Hawkey et al., 2015; Keane et al., 2013). Furthermore, most of tools have been developed to detect TE in the genomes of eukaryotes rather than in prokaryotes. Hence, there is still a need for a simple tool to identify insertions in bacterial genomes using structural variant detection method from short-read data, but which export data that facilitate the validation of IS insertions ab initio (i.e. with a database-free approach).
Here, we created a tool that identifies and locates unknown IS insertion in bacterial genomes from NGS data. We evaluated its sensitivity and precision on simulated data, compared its performances with those of available tools and used it to decipher the evolution of a high-risk clone lineage of the pathogenic species Pseudomonas aeruginosa.
2 Materials and methods
2.1 Software design
The panISa program is a python script that parses a read-mapping file SAM/BAM (Li et al., 2009) using the PYSAM library (https://github.com/pysam-developers/pysam). Potential IR regions are detected with EINVERTED executable from the EMBOSS package (Rice et al., 2000). PanISa was developed under the GPL3 license and is freely accessible at Github (https://github.com/bvalot/panISa). We named it panISa, with pan- as a prefix (Greek pan, ‘all’) and IS for IS, making a pun with the first author’s name.
2.2 Evaluation of the panISa performances
We evaluated the performance of panISa using the genomes of five major human bacterial pathogens: Escherichia coli, Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus and Vibrio cholerae, in which ISs were artificially inserted. Sequence data from reference strains and ISs were obtained from NCBI and ISFinder, respectively (Table 1 and Supplementary Table S1; Siguier et al., 2006).
Reference bacterial strains used to evaluate the panISa program
| Strains | NCBI accession number | ISa (n) |
|---|---|---|
| Escherichia coli K12 substrain MG1655 | NC_000913 | 22 |
| Mycobacterium tuberculosis H37Rv | NC_000962 | 3 |
| Pseudomonas aeruginosa PAO1 | NC_002516 | 25 |
| Staphylococcus aureus NCTC8325 | NC_007795 | 8 |
| Vibrio cholerae O1 biovar El Tor strain N16961 | NC_002505 and NC_002506b | 9 |
| Strains | NCBI accession number | ISa (n) |
|---|---|---|
| Escherichia coli K12 substrain MG1655 | NC_000913 | 22 |
| Mycobacterium tuberculosis H37Rv | NC_000962 | 3 |
| Pseudomonas aeruginosa PAO1 | NC_002516 | 25 |
| Staphylococcus aureus NCTC8325 | NC_007795 | 8 |
| Vibrio cholerae O1 biovar El Tor strain N16961 | NC_002505 and NC_002506b | 9 |
aNumber of different ISs used in simulation (Supplementary Table S1 for details).
bThe two NCBI accession numbers correspond to the two chromosomes of the strain.
Reference bacterial strains used to evaluate the panISa program
| Strains | NCBI accession number | ISa (n) |
|---|---|---|
| Escherichia coli K12 substrain MG1655 | NC_000913 | 22 |
| Mycobacterium tuberculosis H37Rv | NC_000962 | 3 |
| Pseudomonas aeruginosa PAO1 | NC_002516 | 25 |
| Staphylococcus aureus NCTC8325 | NC_007795 | 8 |
| Vibrio cholerae O1 biovar El Tor strain N16961 | NC_002505 and NC_002506b | 9 |
| Strains | NCBI accession number | ISa (n) |
|---|---|---|
| Escherichia coli K12 substrain MG1655 | NC_000913 | 22 |
| Mycobacterium tuberculosis H37Rv | NC_000962 | 3 |
| Pseudomonas aeruginosa PAO1 | NC_002516 | 25 |
| Staphylococcus aureus NCTC8325 | NC_007795 | 8 |
| Vibrio cholerae O1 biovar El Tor strain N16961 | NC_002505 and NC_002506b | 9 |
aNumber of different ISs used in simulation (Supplementary Table S1 for details).
bThe two NCBI accession numbers correspond to the two chromosomes of the strain.
For simulation, ISs were randomly inserted in the genome along with a DR, of which the length was consistent with those of the IS family. Thirty ISs were randomly picked for each bacterial species, with at least one representative of each IS family. Illumina short reads were simulated from each ‘artificial’ genome using DWGSIM v.0.1.11-3 (https://github.com/nh13/DWGSIM) and aligned against the original genome using BWA-MEM (Li and Durbin, 2009). We ran panISa on the aligned reads to detect the IS and compared the output with the expected results.
We evaluated the impact of read-data quality on the performance of panISa by simulating three read lengths (100, 150 and 300 bp) and five coverage depths (20, 40, 60, 80 and 100x) for each bacterial genome (1 genome per species except 2 for V. cholerae) in which 30 ISs were inserted. Each point was repeated ten times, resulting in a total of 27 000 simulated IS transpositions. The factors influencing sensitivity and precision of the detection were analyzed by ANOVA, followed by Tukey’s range test for those which were significant. Results were considered to be significant for P-values < 0.01.
2.3 Analysis of Pseudomonas aeruginosa clinical isolates
Five clinical isolates of P. aeruginosa were collected from patients hospitalized in the University Hospital of Besançon (France) from October 2007 to May 2008. They were considered to be clonal since they shared the same band pattern after pulsed-field gel electrophoresis (data not shown; Talon et al., 1996). Multilocus sequence typing revealed that they all belonged to the high-risk clone ST233. After DNA extraction, genome isolates were sequenced using Illumina HiSeq technology with 2 x150 bp and sub-sampling to 80x by random selection of paired-end reads. The genome of the earliest isolate (10–2007) was assembled using Ray software with the default value for Illumina data (Boisvert et al., 2010), generating 118 contigs for a total of 6,997,480 bp, from which 5086 genes were detected by Prodigal (Hyatt et al., 2010). We then used blastP to search for homologous proteins against the proteome of P. aeruginosa (Uniprot, 50 812 entries). The raw WGS data of the four later isolates (11–2007, 01–2008, 04–2008 and 05–2008) were aligned against the assemblies of the 10–2007 isolate using BWA-MEM (Li and Durbin, 2009) and variants were called using Freebayes (https://github.com/ekg/freebayes). We experimentally assessed the mutation rate of all clinical isolates and that of the PAO1 reference strain using the procedure previously described (Oliver et al., 2002). ISs were detected by running panISa on the five aligned ST233 genomes with the ‘minimum clipped reads’ option set at 20. Left and right sequences of potential ISs were clustered using Sumaclust (https://git.metabarcoding.org/obitools/sumaclust/wikis/home) with an identity of 95%. We then searched for homologous sequences for each representative in the ISFinder database (Siguier et al., 2006). The presence and position of IS sequences was further experimentally verified by PCR and sequencing with specific primers (Supplementary Table S2).
3 Results and discussion
3.1 The regular NGS analysis pipeline does not reveal IS
We explored the evolution of an epidemic clone of P. aeruginosa ST233 by WGS and retrieved 397 SNPs or indels in the collection with almost all (n = 389) occurring within the genome of the 11–2007 isolate. In contrast, only 7 to 12 SNPs or indels were found in the other genomes. Mutations in 11–2007 were evenly distributed throughout its genome, making a unique horizontal transfer event unlikely. All isolates had been collected within a period of 8 months. Such rapid genetic drift led us to hypothesize that the 11–2007 isolate had a hypermutator phenotype (Oliver et al., 2000). We experimentally found that the 10–2007, 01–2008, 04–2008 and 05–2008 isolates had mutation rates between 2 x 10–8 and 5 x 10–9 (mean, 1.5 x 10–8), comparable to that of the PAO1 reference strain (5.9 x 10–9). In contrast, the 11–2007 isolate had a 159-fold higher mutation frequency (2.4 x 10–6), typical of hypermutators. This phenotype is known to be most often due to mutation in the DNA mismatch repair genes mutS, mutL and uvrD (Oliver et al., 2002). However, mapping of the 11–2007 reads on the 10–2007 assemblies did not reveal any mutations in these three genes. We then assembled the reads of the 11–2007 isolates de novo and carefully searched the DNA mismatch repair genes, revealing that mutS was split between two contigs which had a partial sequence that aligned with ISPa1635 (GenBank accession number AY539834) at one extremity. PCR experiments and sequencing confirmed the ISPa1635 insertion within mutS (Antonio Oliver, personal data). Based on this experience, we aimed to develop a tool that can detect ISs in bacterial genomes from WGS sequencing data using an IS database-free approach.
3.2 Implementation
Careful examination of the alignment of the 11–2007 reads on the 10–2007 assemblies revealed a pattern at the insertion site: many clipped reads (i.e. reads mapping partially on the reference) ending at one side of the DR region (Fig. 1). Most IS detection tools use discordant mate-paired reads combined with genome annotation of repeat regions or a list of known ISs as inputs (Ewing, 2015). The position of the IS is then refined using the clipped read information. Our strategy differs, as the detection of potential IS insertions only relies on clipped reads. The panISa program selects clipped reads and groups them by position and side (start or end) of the clip (Fig. 1B). Two close genomic positions with clipped reads in opposite directions correspond to the boundaries of a potential IS. PanISa then creates the consensus sequence of the DR and the left (IRL) and right (IRR) limits of the inserted sequences from the clipped reads. IRs, which are good markers of ISs, are searched for in IRL and IRR (Siguier et al., 2014). All potential IS insertions are reported in a tabular format (Supplementary Table S3).
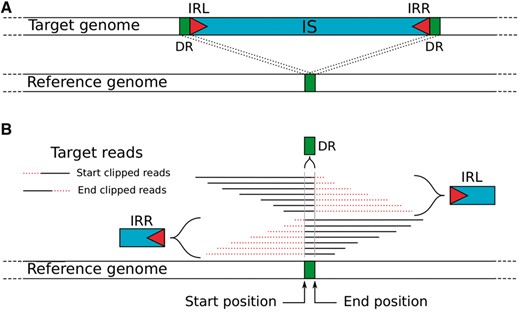
Schematic representation of an IS insertion signature. (A) Structure of the IS insert in the target genome relative to the reference genome. (B) Read alignment of the target genome that partially maps with the reference genome at the IS insertion site. The IRL, IRR and DR could be obtained from these clipped reads
PanISa is designed to potentially detect the insertion of all mobile elements (e.g. ISs, bacteriophages, integrative conjugative elements) or chromosomal rearrangements (i.e. inversions, duplications). The list of potential ISs must be manually inspected and confirmed. The homology of potential ISs with already described ISs is sought by the alignment of the reconstruction of IRL and IRR against the ISFinder database (Siguier et al., 2006). We propose the automation of this validation step with the script ISFinder_search.py (found in the github repository).
3.3 Validation of panISa on simulated data
We evaluated the performance of panISa by artificially inserting ISs in the genomes of reference strains from five major pathogenic bacterial species and assessed the impact of sequencing data quality on the output (i.e. read length and coverage depth). In total, 900 genomes containing 30 IS insertions were simulated. The sensitivity of IS detection depended on the species considered (p = 1.8 x 10–3) and the coverage depth (p = 1.8 x 10–9), but was not affected by the read length (Fig. 2A). The nature of the bacterial species had a slight effect on the sensitivity of our tool. For example, only ISs from P. aeruginosa were significantly retrieved at a higher rate than those of M. tuberculosis, corresponding to the two extremes. In addition, the sensitivity was significantly lower for 20x coverage than for higher coverage (Fig. 2A). For higher coverages (from 40x to 100x), the sensitivity of panISa reached a mean of 98% (95% CI [97.9%–98.2%]).
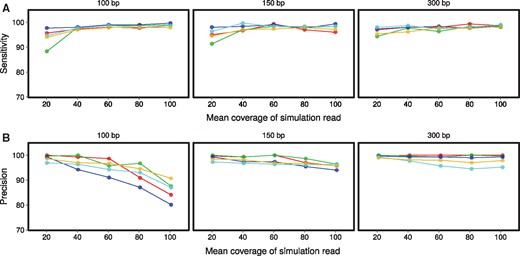
Sensitivity (A) and precision (B) of panISa for IS detection on simulated data. Simulations were performed with read lengths of 100, 150 and 300 bp. Data from E. coli are in red, M. tuberculosis in green, P. aeruginosa in dark blue, S. aureus in light blue and V. cholerae in yellow
The precision of IS detection was affected by read length (p = 4.3 x 10–8) and coverage (p = 4 x 10–7), but not the species considered (Fig. 2B). Multiple comparisons show that the use of 100-bp reads reduced the precision relative to that obtained with 150- and 300-pb reads. Coverage and precision were inversely correlated, particularly with 100-bp reads, for which high coverages (>80x) resulted in lower precision than for a coverage depth of <60x (Fig. 2B). Nevertheless, the precision of the panISa program reached a mean of 98% (95% CI [97.6%–98.6%]) with 150- and 300-pb reads, irrespective of the coverage tested. Most of the false positive positions corresponded to small repeated regions. Hence, more clipped reads of low quality were recognized in these genomic regions with higher coverage depth. This limitation can be easily resolved by sub-sampling high-coverage sequencing data or increasing the minimum number of required clipped reads. In conclusion, the optimal input data for panISa are 150-300-bp reads with a 40 to 60x coverage depth, which allows the detection of ISs with high sensitivity and precision.
3.4 Localization and identification of ISs
The panISa program can determine the position and sequence of DRs and the presence of IRs between the left and right sequences of the IS. PanISa retrieved the exact position and length of the DR in 56.8% of the cases (95% CI [55.5%–57.5%]) from the simulated data. Most (99.5%) of the incorrect predicted DRs were 1- to 5-bp longer than the simulated DR. This error was due to the identity of the nucleotide sequence between the IR and that of the opposite flanking region of the DR, thereby shifting the position of the clip. IRs were detected with a sensitivity of 74% (95% CI [71.2%–76.8%]) and a precision of 99.9% (95% CI [99.87%–100%]). This relatively low sensitivity can be explained by the presence of ISs with highly divergent IRs in the dataset that were never detected (e.g. ISVch7 and ISPa61).
3.5 IS burst in a P. aeruginosa ST233 lineage
The panISa program retrieved 46 new potential insertions in the collection of four late isolates of the epidemic clone ST233 relative to the first isolate, 10–2007. IRL and IRR sequences clustered within eight different sequences, among which six were highly similar to ISs already present in ISFinder (Siguier et al., 2006). These six ISs correspond to 44 mutational events in the genomes of the four late isolates (Table 2). Two putative inserted sequences (one 18-bp insertion and one palindromic region) were manually removed. We experimentally confirmed the IS insertions by PCR with six pairs of specific primers that target the surrounding positions predicted by panISa, using the 10–2007 early isolate as a control (Supplementary Fig. S1). The six PCRs amplified 250–450 bp DNA fragments in the control. Five PCRs amplified DNA fragments that were 1500 to 2500-bp heavier in the genomes of the late isolates, in which ISs were predicted, than in the control. The PCR that targeted ‘ISPpu7 partial’ amplified DNA fragments of similar length in the control and the late isolate in which this IS was predicted. We confirmed the identities of the ISs by sequencing and identified two known ISs (ISPa1635 and ISPa45) and three new ISs (ISPa77, ISPst3 and ISPst5) not yet described in P. aeruginosa (data not shown). Overall, 43 of the 46 IS insertions predicted by panISa were experimentally confirmed. This precision (93.5%) is consistent with that obtained in simulations.
Detection of new ISs in a collection of clonally-related P. aeruginosa ST233 by panISa
| Homologous IS | IS present in the earliest isolate (10‐2007) | Number of new insertions of each IS in late isolates | |||
|---|---|---|---|---|---|
| 11–2007 | 01–2008 | 04–2008 | 05–2008 | ||
| ISPa1635 | Yes | 34a | –– | –– | 2 |
| ISPa45 | Yes | –– | –– | –– | 1a |
| ISPa77 | Yes | –– | 2 | –– | 1a |
| ISPst3 | Yes | –– | 2a | –– | –– |
| ISPpu7 partial | NAb | –– | 1 | –– | –– |
| ISPst5 | Yes | –– | 1a | –– | –– |
| Homologous IS | IS present in the earliest isolate (10‐2007) | Number of new insertions of each IS in late isolates | |||
|---|---|---|---|---|---|
| 11–2007 | 01–2008 | 04–2008 | 05–2008 | ||
| ISPa1635 | Yes | 34a | –– | –– | 2 |
| ISPa45 | Yes | –– | –– | –– | 1a |
| ISPa77 | Yes | –– | 2 | –– | 1a |
| ISPst3 | Yes | –– | 2a | –– | –– |
| ISPpu7 partial | NAb | –– | 1 | –– | –– |
| ISPst5 | Yes | –– | 1a | –– | –– |
aThe presence of a copy of the IS was confirmed by PCR and sequencing (Supplementary Figure S1).
bNot determined.
Detection of new ISs in a collection of clonally-related P. aeruginosa ST233 by panISa
| Homologous IS | IS present in the earliest isolate (10‐2007) | Number of new insertions of each IS in late isolates | |||
|---|---|---|---|---|---|
| 11–2007 | 01–2008 | 04–2008 | 05–2008 | ||
| ISPa1635 | Yes | 34a | –– | –– | 2 |
| ISPa45 | Yes | –– | –– | –– | 1a |
| ISPa77 | Yes | –– | 2 | –– | 1a |
| ISPst3 | Yes | –– | 2a | –– | –– |
| ISPpu7 partial | NAb | –– | 1 | –– | –– |
| ISPst5 | Yes | –– | 1a | –– | –– |
| Homologous IS | IS present in the earliest isolate (10‐2007) | Number of new insertions of each IS in late isolates | |||
|---|---|---|---|---|---|
| 11–2007 | 01–2008 | 04–2008 | 05–2008 | ||
| ISPa1635 | Yes | 34a | –– | –– | 2 |
| ISPa45 | Yes | –– | –– | –– | 1a |
| ISPa77 | Yes | –– | 2 | –– | 1a |
| ISPst3 | Yes | –– | 2a | –– | –– |
| ISPpu7 partial | NAb | –– | 1 | –– | –– |
| ISPst5 | Yes | –– | 1a | –– | –– |
aThe presence of a copy of the IS was confirmed by PCR and sequencing (Supplementary Figure S1).
bNot determined.
The five ISs predicted in silico were already in the genome of the earliest isolate (10–2007) of the epidemic clone P. aeruginosa ST233. During the spread of this clone in hospitalized patients, the ISs mostly duplicated once or twice in new locations of the chromosomes of the late isolates (Table 2). In contrast, ISPa1635 proliferated in the hypermutator isolate (11–2007), in which we found 34 copies (Fig. 3), presumably due to the mutation of the DNA mismatch repair gene mutS (Oliver et al., 2002).
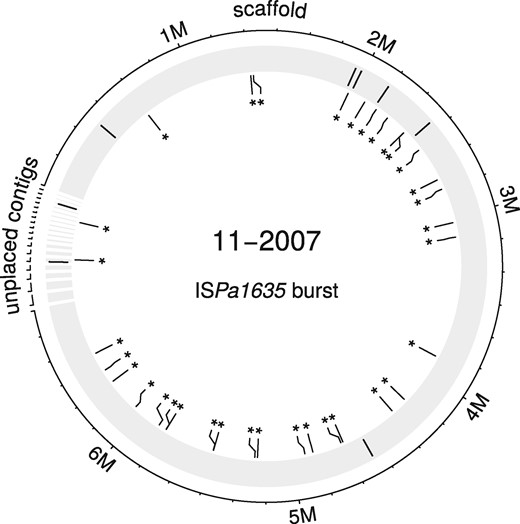
IS burst in a P. aeruginosa ST233 lineage. Initial positions of ISPa1635 in the chromosome of the early isolate 10–2007 are indicated by black lines on the shaded outer circle. New insertions of the ISPa1635 in the late isolate 11–2007 are represented by asterisks
3.6 Comparison with existing IS detection tools
The performances of panISa were compared to those of existing tools (Table 3) on the sequencing data of the isolate 11–2007 using the genome of the 10–2007 isolate as a reference.
Comparison of different tools for the detection of ISPa1635 burst in the P. aeruginosa isolate 11–2007
| Tool | Type of detection | Super kingdom targeted | Number of ISPa1635 insertions detected | Number of false positivea |
|---|---|---|---|---|
| PanISa | Structural variant/ TE detection | Prokaryote | 34 | 1 |
| breseq | Structural variant | Prokaryote | 32 (2)b | 6 |
| Mantac | Structural variant | Eukaryote | 0 | ND |
| Pindel (DD_detection) | Structural variant | Eukaryote | 15 | ND |
| TIDDIT | Structural variant | Eukaryote | 32 | ND |
| ISMapper | TE detection | Prokaryote | 34 | 1 |
| RetroSeq | TE detection | Eukaryote | 0 | ND |
| RelocaTE | TE detection | Eukaryote | 30 | 4 |
| TE-Located | TE detection | Eukaryote | 32 | 17 |
| Tool | Type of detection | Super kingdom targeted | Number of ISPa1635 insertions detected | Number of false positivea |
|---|---|---|---|---|
| PanISa | Structural variant/ TE detection | Prokaryote | 34 | 1 |
| breseq | Structural variant | Prokaryote | 32 (2)b | 6 |
| Mantac | Structural variant | Eukaryote | 0 | ND |
| Pindel (DD_detection) | Structural variant | Eukaryote | 15 | ND |
| TIDDIT | Structural variant | Eukaryote | 32 | ND |
| ISMapper | TE detection | Prokaryote | 34 | 1 |
| RetroSeq | TE detection | Eukaryote | 0 | ND |
| RelocaTE | TE detection | Eukaryote | 30 | 4 |
| TE-Located | TE detection | Eukaryote | 32 | 17 |
aFalse positives were evaluated only for tools which give IS insertion or junction with known IS regions; ND, not determined.
bbreseq identified IS insertion where two new junctions were connected with ISPa1635 in reference genome. The number in parenthesis indicates the number of cases where only one side junction was found.
cManta could not be run in haploid mode, as this parameter is not in option.
dTE-Locate found most of the insertions of ISPa1635, but all positions were shifted from 400 to 600 bp.
Comparison of different tools for the detection of ISPa1635 burst in the P. aeruginosa isolate 11–2007
| Tool | Type of detection | Super kingdom targeted | Number of ISPa1635 insertions detected | Number of false positivea |
|---|---|---|---|---|
| PanISa | Structural variant/ TE detection | Prokaryote | 34 | 1 |
| breseq | Structural variant | Prokaryote | 32 (2)b | 6 |
| Mantac | Structural variant | Eukaryote | 0 | ND |
| Pindel (DD_detection) | Structural variant | Eukaryote | 15 | ND |
| TIDDIT | Structural variant | Eukaryote | 32 | ND |
| ISMapper | TE detection | Prokaryote | 34 | 1 |
| RetroSeq | TE detection | Eukaryote | 0 | ND |
| RelocaTE | TE detection | Eukaryote | 30 | 4 |
| TE-Located | TE detection | Eukaryote | 32 | 17 |
| Tool | Type of detection | Super kingdom targeted | Number of ISPa1635 insertions detected | Number of false positivea |
|---|---|---|---|---|
| PanISa | Structural variant/ TE detection | Prokaryote | 34 | 1 |
| breseq | Structural variant | Prokaryote | 32 (2)b | 6 |
| Mantac | Structural variant | Eukaryote | 0 | ND |
| Pindel (DD_detection) | Structural variant | Eukaryote | 15 | ND |
| TIDDIT | Structural variant | Eukaryote | 32 | ND |
| ISMapper | TE detection | Prokaryote | 34 | 1 |
| RetroSeq | TE detection | Eukaryote | 0 | ND |
| RelocaTE | TE detection | Eukaryote | 30 | 4 |
| TE-Located | TE detection | Eukaryote | 32 | 17 |
aFalse positives were evaluated only for tools which give IS insertion or junction with known IS regions; ND, not determined.
bbreseq identified IS insertion where two new junctions were connected with ISPa1635 in reference genome. The number in parenthesis indicates the number of cases where only one side junction was found.
cManta could not be run in haploid mode, as this parameter is not in option.
dTE-Locate found most of the insertions of ISPa1635, but all positions were shifted from 400 to 600 bp.
We first tested four tools searching for structural variant: breseq dedicated to prokaryotes (Barrick et al., 2014), Manta, Pindel (DD_detection) and TIDDIT dedicated to eukaryotes (Chen et al., 2016; Eisfeldt et al., 2017; Kroon et al., 2016). We ran breseq with the annotation of the IS in the reference genome. This tool correctly located the 34 ISs, although 2 ISs were found with only a single junction. Manta found no breakpoints at insertion sites of ISs, probably because the program could not be run in haploid mode. Pindel reported all discordant pair-end reads, but only retrieved 15 of the 34 IS locations with sufficient accuracy. TIDDIT found 32 positions with a break end.
We then tested four TE detection tools: ISMapper developed for prokaryotes (Hawkey et al., 2015) and RetroSeq, RelocaTE and TE-Locate developed for eukaryotes using McClintock pipeline (Keane et al., 2013; Nelson et al., 2017; Platzer et al., 2012; Robb et al., 2013). All tools were run on the 11–2007 isolate using the 5 ISs sequences identified by panISa (ISPa1635, ISPa45, ISPa77, ISPst3 and ISPst5) as input. RetroSeq retrieved no IS. RelocaTE and TE-Locate detected IS insertions at 30 and 32 positions, respectively. Suprisingly, all positions detected by TE-Locate were shifted of around 400–600 bp. ISMapper retrieved the same 34 ISs than panISa. False positive results were mostly due to similarity with genes encoding transposase or presence of large deletions in the genome.
Overall, all tools optimized for prokaryotes found the same 34 positions. However, breseq and ISMapper needed the position of IS in the reference genome and the sequence of IS to search as input, respectively. In contrast, tools dedicated to eukaryotes produced highly heterogeneous results. Hence, some tools retrieved the majority of the IS positions while other only gave a negative signal.
4 Conclusion
IS elements can deeply affect the evolution of their host (Vandecraen et al., 2017). However, this role is presumably underestimated, since the detection of ISs from high-throughput short-read sequencing data is complicated. Available tools are restricted to the detection of known ISs, preventing the identification of new elements (Ewing, 2015). Here, we developed panISa, a sensitive and precise program for the ab initio detection of ISs in bacterial genomes. PanISa only requires short reads and a reference sequence as input. We validated this new tool on the genomes of five major human bacterial pathogens using simulated data from the widespread technology Illumina. We explored the evolution of a lineage of the high-risk clone of P. aeruginosa ST233 with panISa. It allowed the identification of the transposition of five different ISs during the spread of this clone among patients and a burst of one IS in a hypermutable isolate. Overall, panISa will accelerate the identification of new ISs from short-read data and will enrich the panel of existing tools for the elucidation of the evolution of prokaryote lineages.
Acknowledgements
The authors thank Antonio Oliver from the Hospital Universitario Son Espases, Instituto de Investigación Sanitaria Illes Balears, Palma de Mallorca, Spain for helpful comments. Computations have been performed at the ‘Mésocentre de Calculs de Franche-Comté’.
Funding
PT was supported by a grant from Prince of Songkla University, Thailand. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}