





Abstract
Many genomic features are defined not by exact sequence matches, but by degenerate nucleotide motifs that represent multiple compatible matches. While there are databases cataloging genomic features, such as the location of transcription factor motifs, for commonly used model species, identifying the locations of novel motifs, known motifs in non-model genomes, or known motifs in personal whole-genomes is difficult. I designed motif scraper to overcome this limitation, allowing for efficient, multiprocessor motif searches in any FASTA file.
The motif scraper package (MIT license) is available via PyPI, and the Python source is available on GitHub at https://github.com/RobersonLab/motif_scraper.
1 Introduction
Genomic features can often be described by sequence motifs, rather than exact sequence matches. Particularly important examples of this property are proximal promoter elements that bind transcription factors, and proteins that bind at enhancers and insulators. In these cases, the binding protein does not find an exact sequence match, but rather binds a range of sequences with compatible charge profiles for the protein binding interface. Using methods such as ChIP-Seq, the binding sequences for these factors can be determined and represented as a sequence motif using IUPAC-approved degenerate nucleotide codes. Some important features are exact matches, such as the match between a microRNA (miR) and seed sequences in the 3′ untranslated region (UTR) of a targeted gene. Others have well-defined degeneracy, such as genome-editing target sites. Many databases exist cataloging the location of transcription factor motifs (Kaplun et al., 2016; Kel et al., 2003; Knuppel et al., 1994; Matys et al., 2006; Wingender et al., 1996; Wingender, 1988; Wingender, 2008), miRNA binding sites (Andres-Leon et al., 2015; Dweep et al., 2014; Griffiths-Jones, 2004; Griffiths-Jones et al., 2006; Griffiths-Jones et al., 2008; Kozomara and Griffiths-Jones, 2014; Lagana et al., 2012; Prabahar and Natarajan, 2017), and genome-editing sites (Gratz et al., 2014; Heigwer et al., 2014; Liu et al., 2015; Montague et al., 2014; Naito et al., 2015; Stemmer et al., 2015; Xiao et al., 2014). However, these databases are often restricted to commonly used model species. Newly sequenced species are likely never to be included, and model species may lag behind the release of new genome drafts. Furthermore, many individual, phased whole genomes are being generated. The databases of sequence motifs are designed relative to a reference sequence, rather than to personal genomes. There are other tools that exist that could identify motifs based on a position-weight matrix or other information, such as ChIP-Seq peaks and DNase hypersensitivity, including HOMER (Heinz et al., 2010) and MEME-Suite (Bailey et al., 2009). The downside of de novo motif identification is often a substantial time trade off.
Inspired by previous work to identify a specific subset of CRISPR/Cas9 sites (Roberson, 2015), my goal for motif scraper was to instead develop a more general purpose motif searching tool that would have broader use. Motif scraper fills this annotation gap by allowing for the specification degenerate sequence motifs and reporting the location and composition of all matches in a FASTA file, which could be a personal genome, a reference genome, or a set of genomic slices, such as all the 3′ UTRs of protein coding genes. This tool therefore functions more as a FASTA degenerate sequence ‘grep’ that is easy to install and use, and scales well with full genome sequence files.
2 Materials and Methods
Motif scraper was designed in Python, and is compatible with both Python 2 and 3. The ability to read FASTA formatted files and generate FASTA indexes is provided by pyfaidx (Shirley et al., 2015). Motifs are specified as a text string with using IUPAC degenerate bases, which are converted internally into a regular expression and compiled by the regex package. This allows for detection of overlapping motifs. One or more specific regions or a specific strand relative to the reference can be specified for targeted search. By default all contigs in the FASTA file are searched for both + and − strands. The multiprocessor Python package handles the use of multiple computer cores, searching each target region/strand separately. Each hit is reported with the contig, start position, end position, strand, sequence, and matching motif in the output file. The code is available under an MIT license, stored on GitHub, and distributed through the Python Package Index (PyPI). Compatibility with Python 2 and 3 is assessed with every repository commit using Travis CI service. This paper used motif scraper v1.0.1.
3 Results
3.1 Identification of mock transcription factor binding motifs
As a benchmark, I calculated a faux consensus sequence for two DNA binding proteins: CCAAT/Enhancer Binding Protein Beta (CEBPB) and CCCTC-Binding Factor (CTCF). I downloaded their Position Weight Matrices for Homo sapiens from Jaspar (Mathelier et al., 2016). I then calculated the fraction of weight at each position attributable to each base. At each position I considered a base contributing at least 5% of overall weight to be a possible match at that position. I then converted these possible base matches per position into degenerate IUPAC bases to form an estimated degenerate motif. The CEBPB (MA0466.2) calculated motif was VTKDYRHAAY, and the CTCF (MA0139.1) calculated motif was NNNMCDSNAGRDGDHRVNN. I also downloaded the MEME formatted position-weight matrix (PWM) for both motifs for use with MEME-Suite. I compiled MEME-Suite v4.12.0 from the source code using gcc/g++v5.4.0. I used the FIMO (FInd MOtif) tool to search the human genome (Ensembl GRCh38 release 91) for binding sites for both motifs with default settings. I tested the performance of multiple processors for the faux motifs using 1–10 processors on a machine with an Intel i7-3929k 3.20 GHz processor and 32 GB RAM running Ubuntu 16.04.1 64-bit and Python 2.7.12. In this benchmark motif scraper had decreased run time with additional processors (saturating at ∼6), and required more time for longer motifs (Fig. 1).
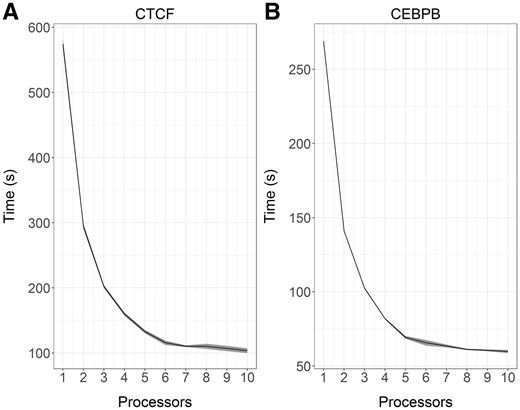
(A, B) Runtimes for variable processor usage. Above are the runtimes for two motifs on the same system using 1–10 processors. The dots represent means, and the ribbons show the standard deviation for ten iterations of each condition
3.2 Comparison to MEME-suite
FIMO is designed to not just identify potential matches to a motif, but also to enrich for potential matches present greater than expected by chance given genomic background. FIMO therefore requires significant computational time. For CEBPB, motif scraper identified 4 568 172 potential sites based on my definition of the binding degeneracy, whereas FIMO found 61 123 significantly enriched binding sites. For CTCF, motif scraper found 496 026 sites and FIMO found 53 566 sites. This highlights the major differences in the tools. FIMO is designed to give you a likely binding site based on the PWMs. The final lists are relatively small and likely to be non-random. However, this operation is slow. For CEBPB, FIMO took 1435.0 s ± 19.8 s to find the enriched sites. Out of the enriched sequences, ATTACACAAT was the most common (10 927/61 123). Searching for that specific sequence with motif scraper using only 1 processor took only 209.6 s ± 0.5 s with 100% overlap with the FIMO. Therefore, for transcription factor binding sites, finding significantly enriched motifs clearly benefits from taking background sequence into context and requires additional computational time. However, for sequences not based on a PWM, motif scraper can significantly decrease processing time.
4 Summary
The lack of portable, general-purpose motif-finding tools for uses such as genome annotation is a significant barrier for the discovery of motifs in new/non-model genomes. The rapid increase in the number of available whole-genomes only amplifies this problem. Motif scraper aims to fill this gap. This tool has cross-platform compatibility and a permissive license for broad reuse. The runtime for annotation of relatively degenerate nucleotide sequences is fast, on the order of minutes for a whole-genome using multiple processors. The FASTA format allows for flexible input, ranging from whole genomes down cDNA sequences and plasmids. It could also be used to search for potential microRNA binding seed sequences in 3′ UTRs to predict potential partners for organisms not available in TargetScan (Agarwal et al., 2015).
It is worth noting that this tool cannot, and does not aim to, replace probabilistic binding models. For interactions best specified by a position-specific weighted matrix, other tools that quantify enrichment over background are more apt. But for sequences that are well-defined and exact, such as restriction enzyme sites and microRNA binding sites, or that have defined degeneracy, such as genome-editing motifs, motif scraper can annotate their location with ease.
It is also worth noting that the performance of parallel processing is highest with few relatively large contigs, i.e. reference genomes. The algorithm can be applied to smaller contigs, such as 3′ UTRs from a whole-genome to identify microRNA binding sites. However, the performance decreases appreciably with many short contigs. This limitation could be overcome by instead processing a batch of contigs per core to limit the number of data transfer operations. Overall, the broad operating system compatibility, use of a standard input format, and relative speed help support motif scraper as an important tool for non-model organisms and annotation of non-standard motifs.
Acknowledgements
Special thanks to Dr. Karyn Meltz Steinberg for her helpful discussions during the development of this tool, and to the reviewers for significantly improving the manuscript.
Funding
This work was partially supported by the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (P30-AR048335).
Conflict of Interest: none declared.
References


{kind=link}