





Abstract
The direct ancestor of the DNA-protein world of today is considered to have been an RNA-peptide world, in which peptides were co-factors of RNA-mediated catalysis and replication. Evidence for these ancestral peptides, from which folded proteins evolved, can be derived even today from regions of local sequence similarity within globally dissimilar folds. One of these is the 45-residue motif common to both folds of the hnRNP K homology (KH) domain.
In a survey of KH domains, we found a third fold that contains the KH motif at its core. This corresponds to the Small Domain of bacterial Ribonucleases G/E and, like type I and type II KH domains, it cannot be related to the others by a single genetic event, providing further support for the KH motif as an ancestral peptide predating folded proteins.
Supplementary data are available at Bioinformatics online.
1 Introduction
Since at least the time of the Last Universal Common Ancestor (LUCA), proteins have been the fundamental catalysts of life (Lupas and Alva, 2017). For their activity, they must assume three-dimensional structures whose topology is referred to as the protein fold and is determined by its amino acid sequence (Anfinsen, 1973). However, a given fold is compatible with many sequences and these are therefore able to change by mutation and natural selection without changing the overall appearance of the protein. Protein folds thus have a much higher evolutionary permanence than protein sequences (Chothia and Lesk, 1986). Nevertheless, they may change over time by a number of genetic events (e.g. circular permutation, insertion/deletion, or substitution of secondary structure elements; Grishin, 2001a) and it is challenging to judge whether proteins of different fold but significant sequence similarity are locally or globally homologous (Lupas and Koretke, 2008).
One such ‘fold change’ puzzle is presented by the hnRNP K homology (KH) domains (Grishin, 2001b). These ancient domains of about 70 residues bind single-stranded nucleic acids and are built around a 45-residue sequence carrying a conserved (I/L/V)IGxxGxx(I/L/V) pattern, which mediates the nucleic-acid binding (Nicastro et al., 2015). Their structure consists of three α-helices packed against a three-stranded β-sheet, but two topologically different forms are observed (Fig. 1a and b): in type I KH domains (KHI) the secondary structure elements adopt a βααββα (aABbcC) topology and the β-sheet is anti-parallel, while in type II (KHII) the topology is αββααβ (DdaABb) and the β-sheet is mixed parallel and anti-parallel (Fig. 1a and b). The common core of the two types (marked bold above) is referred to as the KH motif. Non-canonical KH motifs, lacking the GxxG pattern, are known and, where tested, lack the ability to bind nucleic acids (Hollingworth et al., 2012).
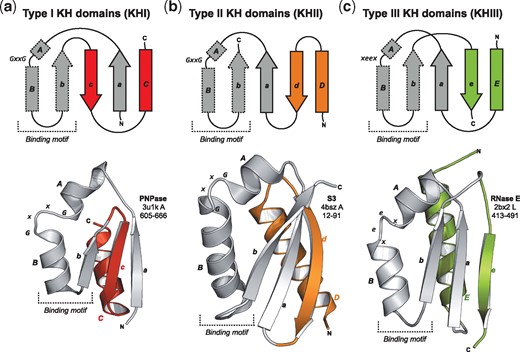
Comparison of KH domain structures. Topology and ribbon diagrams of (a) KHI from human mitochondrial PNPase, (b) KHII from yeast rpS3, and (c) KHIII from the ‘SD’ of Eschirichia coli RNase E with helix F removed for clarity. The KH motif is shown in grey; the different decorations are highlighted
Both folds can be traced to the time of LUCA, but no single genetic event can be conceived to explain their evolutionary connection (Grishin, 2001b; Lupas et al., 2001). It is thus difficult to rationalize how one of the KH forms originated from the other by a chain of events that, at a minimum, must have included accretion, strand displacement and deletion. For this reason, an alternative scenario was considered already at the time when the KH motif was described; in this, the motif corresponded to a peptide active in the RNA-peptide world and was later independently decorated at each terminus to yield two different folds. This view received support from the recent reconstruction of a set of ancestral fragments, which may represent the last observable remnants of these peptides; this set included the KH motif (Alva et al., 2015). Indeed, the activity of the KH motif in binding single-stranded nucleic acids would have been highly relevant for an RNA-based life form. In an effort to gain more insight into the origins of these domains, we made a survey of proteins carrying KH matches. These searches were limited to the prokaryotic lineages in order to focus on the time around LUCA and revealed the existence of yet another type of KH domain with a distinct topology, supporting a fragment-based view of KH domain origin.
2 Materials and methods
2.1 Identification of KH-containing proteins
We used a two-step procedure to detect proteins containing a KH match, building initial profiles from proteins of known structure and refining these separately against the UniProt Reference Proteomes of archaea and bacteria as of June 2017. KHI and KHII sequences were retrieved from the SCOPe superfamily d.51.1 and the family d.52.3.1, respectively (Fox et al., 2014). The sequences from Polyribonucleotide nucleotidyltransferase (PNPase), the single KH incorrectly annotated, were removed from the KHII set and the sequences of KHII domains in the PDB not yet assigned to a SCOPe family were added (Supplementary Table S1). Each set was aligned with PROMALS3D (Pei and Grishin, 2014) and used for HMM searches with HMMER (Finn et al., 2015), which allowed the identification of protein families carrying complete or partial KH sequences. The domain boundaries suggested by the searches were checked with HHpred (Söding et al., 2005) over SCOPe95 using the MPI Bioinformatics Toolkit (Zimmermann et al., 2018).
2.2 Classification and sequence comparison of KH-containing domains
The obtained matches were clustered with CLANS (Frickey and Lupas, 2004) based on their BLASTp P-values (Supplementary Fig. S1) and the individual clusters were then enriched with sequences from the reference proteomes, using HMM comparisons. For those cases where the domain boundaries suggested by the HMMER searches agreed with the HHpred searches, the matches from the corresponding clusters were aligned and used for iterative searches, as described further below. For the two cases in which they did not agree [archaeal ATPase PINA and bacterial ribonucleases (RNases) E/G] we defined domain boundaries and collected their sequences based on the structure of the matched regions.
For PINA, the secondary structure of the matched region in the proteins from Methanocaldococcus jannaschii (UniProtKB: Q58928) and Sulfolobus islandicus (UniProtKB: C3MQK6) was predicted with Quick2D (Alva et al., 2016) and its three-dimensional structure generated template-free with RaptorX Contact Prediction (Wang et al., 2017; Supplementary Fig. S2). For RNases G/E, the matches were mapped onto the automatically re-refined (Joosten et al., 2009) structure of E. coli RNase E (PDBid: 2xb2 L; Supplementary Fig. S3a and b) and regions homologous to the matched domain in other proteins identified with HHpred (Supplementary Fig. S4). Alignments for these were added to those derived above.
Each alignment was used for iterative searches over the reference proteomes with JackHMMER (Finn et al., 2015) until convergence. Each search started with significance E-values of 1 x 10−10 for the sequence and 3 x 10−10 for the hit, and was monitored for the inclusion of spurious matches. The resulting matches were filtered to a maximum sequence identity of 70% with CD-HIT (Li and Godzik, 2006). The final 9.739 KH-like sequences and the 1.180 sequences distantly related to the RNase E/G matches were used to build the final cluster maps with CLANS (Fig. 2a and Supplementary Fig. S4). The same clusters were obtained as in the first step of sequence searches; their sequence space, however, is now better represented, covering a total of 168 archaeal and 2059 bacterial proteomes. HMM sequence profiles were built for each cluster with hhmake and aligned with hhalign (Fig. 3; Söding, 2005). For that, each cluster was aligned and processed with trimAL (Capella-Gutierrez et al., 2009) by removing columns where more than 85% of the positions represent a gap (gap score of 0.15) and sequences that only overlap with less than 50% of the columns populated by 80% or more of the other sequences. HMM profiles were calculated without secondary structure scoring.
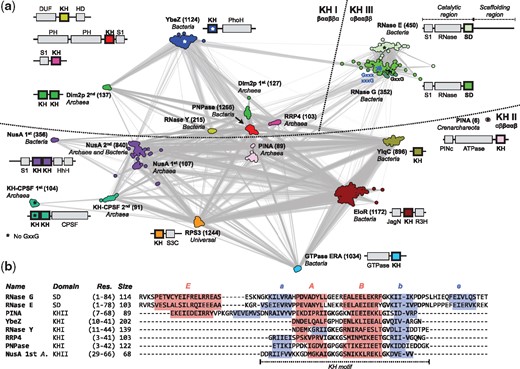
Classification and alignment of prokaryotic KH and KH-like sequences. (a) Cluster map of KH-containing domain sequences. Clustering was done in 2D until equilibrium at a BLASTp P-value of 10–14. The number of sequences in each cluster is shown in brackets. Connections represent similarities up to a P-value of 10–10 (darker means more similar). First and second indicate different KH domains within the same protein. (b) Alignment of the consensus sequences from selected clusters, obtained by HMM comparison to the SDs from RNase G. Predicted strands and helices are highlighted. The number of sequences (size) automatically selected by hhmake to build the HMM profile is shown
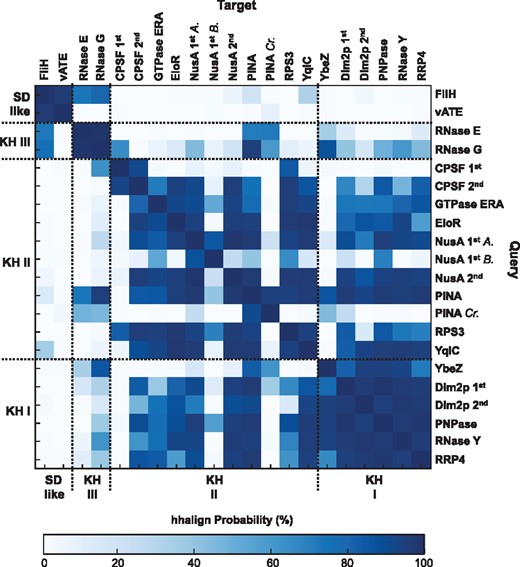
HMM-HMM comparison of KH, KH-like and SD-like sequences. A.: Archaea; B.: Bacteria; Cr.: Crenarchareota
3 Results and discussion
3.1 Protein families with KH-like matches
We identified KHI and KHII matches from 10 prokaryotic families known to carry at least one of these domains (Supplementary Table S2 and Fig. 2a). Based on the taxonomic distribution and the internal organization of their clusters, two families can be traced to LUCA: the KHII in ribosomal protein S3 and the 2nd KHII from transcription termination/anti-termination protein NusA. Additionally, we identified two further families containing KH domains not hitherto reported (Supplementary Table S2 and Fig. 2a). Of these 12 families, three contain a non-canonical KH motif: bacterial YbeZ, archaeal KH-CPSF and crenarchaeal PINA (far separated from other archaeal PINA sequences in the cluster map but predicted to adopt a KHII fold, Supplementary Fig. S2).
Unexpectedly, we obtained closely connected clusters for matches from bacterial RNases G/E (Figs 2a and 3), also found in chloroplasts and not hitherto reported to carry a KH domain (Aït-Bara et al., 2015). RNase G is the oldest member of this family; it is found in the proteomes of several eubacteria and is involved in the maturation of ribosomal RNA and the degradation of transcripts. It forms a dimer and is composed by an S1 RNA-binding domain, an RNase domain and a ‘Small Domain’ (SD; Fig. 2a). RNase E is involved in the processing and degradation of transcripts in proteobacteria and is an important component of their degradosome. It forms a tetramer and has an N-terminal catalytic region homologous to RNase G (Fig. 2a).
The only crystallographic structure known for this family is that of the catalytic region of E. coli RNase E; when mapped onto this, the KH-like region falls within the 75-residue SD. SDs in all RNases E and most RNases G lack the GxxG pattern. The pattern is present in RNases G from armatimonadetes, actinobacteria and firmicutes (Supplementary Fig. S6), especially clostridia; partial patterns (Gxxx and xxxG) were found in RNases G from several bacterial lineages, including α-, γ- and δ-proteobacteria (Supplementary Fig. S6). Thus, SD sequences are connected to KH domains via RNases G (Fig. 2a), mainly via those from clostridia. They are close to the KHII from archaeal PINA and the KHI from bacterial YbeZ (P ≥ 80%; Fig. 3).
3.2 Shared features of KH and SD domains
The SD is not classified in CATH or SCOPe, and forms a clade by itself as ‘Thioredoxin-like domain in RNase E’ within the ‘a + b three layers’ category of ECOD (Cheng et al., 2014). Here, a mixed three-stranded β-sheet is packed between four α-helices (Supplementary Fig. S3a), resembling KHI and KHII folds (Fig. 1c). Its secondary structure elements, however, follow an αβααββα (EaABbeF) topology, with the match to KH domains located in the central βααβ fragment, especially the core ααβ (ABb) elements (Fig. 2b). This region is essential for RNase E tetramerization, forming the core of the SD-SD dimer interface (Supplementary Fig. S3d); structurally, it aligns to canonical KH motifs with an average TMscore of 0.47 ± 0.05, which is comparable to that of other non-canonical KH motifs (Supplementary Fig. S5).
The SD of E. coli RNase G lacks the C-terminal helix (F) and preceding loop of RNase E SD (Supplementary Fig. 3c); this may explain why E. coli RNase G is a dimer. As this region is not conserved in other RNases G/E (Supplementary Fig. S7), we consider the SD topology to be αβααββ (EaABbe). A similar topology is observed in the globular domains of the flagellum-specific ATP synthase FliH and the subunit E of the V-type ATP synthase (vATE) (Supplementary Fig. S4). These may be distantly related to the SD but do not share significant sequence similarity to KHI and KHII domains (Fig. 3).
4 Conclusion
Our results suggest that the SD from RNases E/G is a member of the KH family and that its fold represents a third KH topology: the KH type III (KHIII; Fig. 1). It is built around the KH motif, as both KHI and KHII, and is unlikely to represent a KHI/KHII hybrid. No significant sequence similarity was found that would support an origin by circular permutation from KHI or KHII. However, one could envisage an evolutionary scenario where the SD descended from a KHII close to that from archaeal PINA, which aligns over its entire length to the SD (Fig. 2b). A deletion in strand d would have prevented its formation and required compensation from a C-terminal extension, which would result in a change of orientation of helix E and a re-optimization of its interactions with the remaining domain. However, the alignment of helix A from PINA with helix E from RNases E/G may be solely a result of their amphipathic nature. Thus, we consider it more plausible that, like KHI and KHII, KHIII evolved independently by decoration of an ancestral peptide represented by the KH motif. However, due to the large time of divergence from LUCA, an evolutionary connection by a chain of complex untraceable events should not be dismissed (Grishin, 2001a).
We only detected SD in bacterial proteins and cannot trace it to the time of LUCA. However, this can be a result of the significantly lower number of archaeal proteomes available. As the SD carries a canonical KH pattern in some bacterial species and is found together with two domains involved in RNA binding and metabolism, particularly S1, which is found in the context of all KH types (Fig. 2a), it is plausible that its non-canonical form evolved from an antecedent KHIII originally able to bind nucleic acids. In this scenario, this form may have lost its function and was positively selected to allow, for example, for RNase G/E oligomerization, originating a new family of protein-interacting domains, possibly further giving rise to the globular domains of FliH and vATE.
Acknowledgements
The authors thank Vikram Alva, Jens Baßler and Laura Weidemman for stimulating discussions and Hongbo Zhu and Murray Coles for advice on the text.
Funding
This work was supported by institutional funds from the Max Planck Society.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}