





Abstract
Genome sequencing has had a remarkable impact on our ability to study the effects of human genetic variation, however, variant interpretation remains the major bottleneck. Understanding the potential impact of variants, including structural variants, requires extensive annotation from disparate sources of knowledge, and in silico prediction algorithms.
We introduce Seave, an intuitive web platform that enables all types of variants to be securely stored, annotated and filtered. Variants are annotated with allele frequencies and pathogenicity assessments from many popular databases and in silico pathogenicity prediction scores. Seave enables filtering of variants with specific inheritance patterns, including somatic variants, by quality, allele frequencies and gene lists which can be curated and saved. Seave was made for whole genome data and is capable of storing and querying copy number and structural variants.
To demo Seave with public data, see https://www.seave.bio. Source code is available at http://code.seave.bio and extensive documentation is available at http://documentation.seave.bio. Seave can be locally installed on an Apache server with PHP and MySQL, or we provide an Amazon Machine Image for quick deployment. For commercial and clinical diagnostic licensing, contact the corresponding author.
Supplementary data are available at Bioinformatics online.
1 Introduction
The rapid adoption of human genome sequencing has made substantial inroads in our understanding of the impact of genetic variation on health and disease (Delaney et al., 2016). Vast catalogues of genetic variants now exist, tens of thousands of which have been unequivocally linked to disease. Through advances in genomic technologies and bioinformatic methodologies, it is feasible to comprehensively identify all classes of genomic variation within an individual’s genome, ranging from single nucleotide variants (SNVs) and short insertions or deletions (Indels), to large copy number variants (CNVs), structural variants (SVs) and mobile element insertions (MEIs), any of which may contribute to their phenotype.
Interpreting the potential impact of any variant is a difficult task (Amendola et al., 2016), and is a major impediment to widespread adoption of genomic medicine. Variant interpretation requires an assessment of data quality, and investigating dozens of resources, including databases of genomic variation in healthy controls, from patients with disease, resources linking genes to phenotype or disease and the latest literature. All of this information must be kept up to date. Interpreting the impact of novel variants can be supported by potentially dozens of in silico pathogenicity scores. CNVs of any size and SVs are important sources of pathogenic variants, and should be considered alongside short variants. Importantly, this genomic complexity must be distilled, and presented in a way which is accessible to all researchers, clinicians and laboratory staff.
To address these challenges, we developed Seave, a web-based variant filtration platform that stores, queries and annotates genomic variation of all sizes. It is designed for clinicians and researchers, primarily for rare disease and cancer, and requires no knowledge of bioinformatics to use.
2 Seave description
2.1 Scope
Seave was designed from the outset to handle whole-genome-sized variant callsets from tens of thousands of patients, and ably supports data from any sized targeted sequencing panel. Seave supports the following classes of genetic variants: SNVs, Indels, CNVs, SVs and runs of homozygosity (ROH), from the nuclear and mitochondrial genomes. Due to the large file sizes from whole-genome sequencing (i.e. ∼5M variants per patient, and >3 Gb compressed VCF files), Seave is designed to automatically receive data generated by production analysis pipelines via an API (Fig. 1).
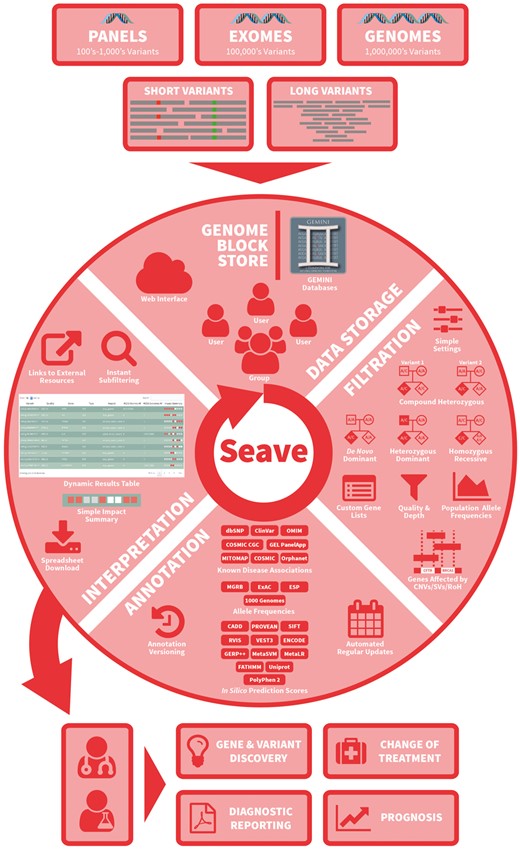
Schematic overview of Seave’s main features. Arrows represent the flow of information through sequencing, variant detection, data storage, filtration, annotation, interpretation and outcomes
2.2 Data import and annotation
Seave uses GEMINI (Paila et al., 2013) databases to store, manage and query genome data, where each database represents a cohort (Supplementary Fig. S1). GEMINI databases are portable, convenient, sharable and allow Seave to scale to vast numbers of individuals by simply increasing disk space. Users can group samples within a cohort into families, and annotate individuals with their gender and affected status, interactively, or via a PED file.
Importing data starts with annotating VCF files containing SNVs and Indels from one or more individuals using Variant Effect Predictor (VEP; McLaren et al., 2016) or SnpEff (Cingolani et al., 2012), then converting these into a GEMINI database. These databases are then imported into Seave using an API, or the administration interface. Seave manages a MySQL annotation database, which complements the annotations from VEP and GEMINI, to provide pre-computed, extensive in silico prediction scores, gene-phenotype-disease links and allele frequencies in healthy controls and various diseases (Fig. 1, Supplementary Data). To keep these annotations up to date, we provide tools for updating those annotations which are regularly updated. Seave supports many popular variant callers including GATK HaplotypeCaller (McKenna et al., 2010), FreeBayes (Garrison and Marth, 2012) and somatic variant callers including Strelka (Saunders et al., 2012), MuTect2 (Cibulskis et al., 2013) and VarDict (Lai et al., 2016). Seave currently supports only the GRCh37 (hg19) reference genome.
To support large CNVs and SVs, we developed the Genome Block Store (GBS). The GBS is a scalable MySQL database designed to store large genomic segments or blocks (e.g. deletions, duplications, inversions, MEIs or ROH), or linked blocks (e.g. gene fusion breakpoints), with additional annotations (e.g. copy number or breakpoint read depth). A number of popular tools are supported, including CNVnator (Abyzov et al., 2011), LUMPY (Layer et al., 2014), Sequenza (Favero et al., 2015), ClinSV (Minoche et al., manuscript in preparation), Manta (Chen et al., 2016), CNVkit (Talevich et al., 2016) and ROHmer (Puttick et al., manuscript in preparation).
2.3 Filtering
After selecting a cohort for analysis (Supplementary Fig. S1), users can optionally select to filter short variants by inheritance pattern, including heterozygous dominant, homozygous recessive, de novo dominant and compound heterozygous (Supplementary Fig. S2). On the Query page, the genomic search space can be restricted or specifically excluded by using any number of genomic coordinates, curated gene lists from Seave’s gene list management system, or custom gene symbols (Supplementary Fig. S3). Variants can be restricted by their impact: low (e.g. synonymous), medium (e.g. missense), high (e.g. nonsense, frameshift and essential splice region), or coding, by CADD score (Kircher et al., 2014) and population allele frequencies. Technical filtering parameters include minimum sequencing depth in all samples, minimum variant quality, excluding failed variants and the type and number of variants to return.
A typical family trio sequenced by whole genome sequencing yields 6 million variants and this number is rapidly reduced to below 200 by just filtering on rarity, impact and inheritance pattern (Supplementary Fig. S4). Most queries take 0–5 s to execute but this can stretch up to 2 min for large cohorts of whole genomes with inheritance patterns specified.
Variants that pass all filters are displayed in a dynamic table, with the extensive annotations noted in Fig. 1 and hyperlinks where applicable (Supplementary Fig. S5). The Impact Summary column visually summarizes the pathogenicity evidence relating to a variant and its cognate gene (Fig. 1, Supplementary Fig. S5). Annotations place variants in the context of the functional genome, and can be dynamically shown using toggle buttons (Supplementary Fig. S6), and sorted by strength of evidence. A unique strength of Seave is that short variants that overlap CNVs or SVs from the same individual in the GBS are highlighted, allowing variants and CNVs to be jointly interrogated. Hyperlinks to control an IGV session are also provided. Results can be shared via their URL, or downloaded to TSV, which includes important auditing information, including timestamps, exact queries used and the versions of all annotations.
There are a number of dedicated queries to interrogate CNVs and SVs, which partially rely on BEDTools (Quinlan and Hall, 2010) to perform interval querying logic. CNVs or SVs can be restricted by gene lists or genomic coordinates, copy number thresholds and minimum CNV size. The SV Fusions search mode is a powerful way to identify candidate gene fusions due to CNV or SV. The Method Overlaps query allows CNVs or SVs identified by multiple callers to be prioritized, whereas the Sample Overlaps query allows CNVs or SVs segregating in families to be prioritized. Finally, the ROHmer query is useful for variant filtering in consanguineous families, and identifies genomic regions of homozygosity that are shared by all affected individuals in a family but not by any unaffected individuals.
2.4 Sharing and security
Seave has a user management system, allowing fine grained data sharing control. Databases are owned by a single group, and users can be members of any number of groups. Administrators can import data, manage users, groups, databases, GBS data and custom gene lists. All login events, data import/export, queries, gene list and user or group changes are audited, and all data is transferred and stored using encryption. Seave is written in PHP to be run on an Apache web server with a MySQL database.
3 Conclusion
Seave was built to enable gene discovery research, diagnostics and precision cancer medicine from whole genome sequences. As a component of Australia’s first clinically accredited whole genome pathology service, Seave has met the rigorous demands of ISO 15189 clinical accreditation. In a research setting, it has been successfully used to discover novel disease genes and variants, as well as rapidly supporting the diagnosis of patients with previously reported pathogenic variants (Balasubramaniam et al., 2017a,b; De Sousa et al., 2017; Ewans et al., 2018; Heimer et al., 2016; Kumar et al., 2016; Riley et al., 2017). Cancer research requires the comprehensive interrogation of large numbers of somatic variants of all sizes and types at differing variant allele frequencies. Accordingly, Seave has been used for characterizing tumour evolution (Merlevede et al., 2016) and as part of two precision cancer genomics programs: the Lions Kids Cancer Genome Project (LKCGP), as part of the Zero Childhood Cancer Program for children with high-risk cancers using whole genome sequencing and for the Molecular Screening and Therapeutics (MoST) program using a targeted genomics screen to test targeted anti-cancer agents in patients with rare or advanced cancers. Finally, Seave has been used for training purposes in Australia and Hong Kong, across multiple clinical genomics data analysis workshops for clinical geneticists, researchers, laboratory scientists and other health professionals.
Acknowledgements
We thank members of the Translational Genomics group (KCCG) and Genome. One for their comments and questions that lead to new features and bug fixes. We also thank Dr. Aaron Quinlan for his creation of GEMINI and generous open source licensing.
Funding
This work was supported by the Kinghorn Foundation, Cancer Institute NSW [grant number 13/ECF/1-46] to MJC and the NSW Department of Health to MJC.
Conflict of Interest: MED is the CEO of Genome.One Pty Ltd, which uses Seave as part of its diagnostic workflow. The remaining authors declare that they have no competing interests.
References


{kind=link}