





Abstract
Understanding the regulatory roles of non-coding genetic variants has become a central goal for interpreting results of genome-wide association studies. The regulatory significance of the variants may be interrogated by assessing their influence on transcription factor binding. We have developed atSNP Search, a comprehensive web database for evaluating motif matches to the human genome with both reference and variant alleles and assessing the overall significance of the variant alterations on the motif matches. Convenient search features, comprehensive search outputs and a useful help menu are key components of atSNP Search. atSNP Search enables convenient interpretation of regulatory variants by statistical significance testing and composite logo plots, which are graphical representations of motif matches with the reference and variant alleles. Existing motif-based regulatory variant discovery tools only consider a limited pool of variants due to storage or other limitations. In contrast, atSNP Search users can test more than 37 billion variant-motif pairs with marginal significance in motif matches or match alteration. Computational evidence from atSNP Search, when combined with experimental validation, may help with the discovery of underlying disease mechanisms.
atSNP Search is freely available at http://atsnp.biostat.wisc.edu.
Supplementary data are available at Bioinformatics online.
1 Introduction
Genome-wide association studies have provided overwhelming evidence that a large number of potential causative genetic variants reside in non-coding regions such as intronic or intergenic regions (Nishizaki and Boyle, 2017). These variants might be involved in a variety of regulatory mechanisms such as transcription factor binding, histone modifications or alternative splicing, and play a key role in disease-specific regulatory networks. Significant efforts have been made to identify such regulatory variants especially in the human genome. Comprehensive web resources such as Haploreg and regulomeDB explore functional annotations in human genome at potential regulatory variants (Boyle et al., 2012; Ward and Kellis, 2016). While epigenomic annotations depend on the availability of experimental data in the relevant experimental systems, annotating and nominating genetic variants for their potential regulatory effect on transcription factor binding can be achieved by in silico calculations (Zuo et al., 2015). In disrupting or enhancing transcription factor binding to DNA, regulatory variants might modulate expression levels of disease genes. While the web resources statistically quantify the transcription factor motif matches with both reference and variant alleles, they only nominate a set of regulatory variants passing stringent thresholds and/or do not consider creation of new binding sites by variants.
We built the atSNP Search web resource motivated by the need for (i) a more comprehensive motif-based discovery of regulatory variants; (ii) statistically well-justified quantification of motif matches and changes in motif matches and (iii) their convenient graphical depiction. All the single nucleotide polymorphisms (SNPs) in dbSNP build 144 for human genome assembly 38 (Sherry et al., 2001) were initially examined against motifs from JASPAR (Mathelier et al., 2014) and ENCODE (Kheradpour and Kellis, 2014) libraries by atSNP (Zuo et al., 2015) testing. atSNP Search currently encompasses the results for all SNP-motif combinations having a P-value for either motif matches with the reference or the SNP allele or variant-led changes in motif matches. These statistical quantifications are supplemented with composite sequence logo plots that depict the motif matches with both alleles. None of the existing web databases or software packages provide such automated visual depiction despite the fact that rapid and automated access to sequence logos enables visual exploration of impact of variants on the motif matches. SNP2TFBS, Raven and OncoCis are existing motif-based web databases that nominate regulatory variants with numerical and graphical summaries (Andersen et al., 2008; Kumar et al., 2017; Perera et al., 2014). The Supplementary Material provides a point-by-point comparison of the atSNP Search to these resources.
2 Database contents
The current version of the atSNP Search provides statistical testing results on 37 141 563 102 variant-motif pairs with P-values <0.05 in either motif matches or the change in motif matches. SNPs with multiple alleles are accommodated by comparing each of the alleles with the corresponding reference allele. The atSNP Search motif collection consists of 205 JASPAR motifs and 2065 ENCODE motifs, which represent 792 unique transcription factors with motif lengths between 5 and 30.
3 Search features
The atSNP Search input form has search options with the following queries: (i) a set of SNP rs numbers (RSIDs), (ii) an RSID with a choice of window size around the variant for an extended search region, (iii) genomic coordinates (iv) a gene symbol with a choice of window size for an extended search region around the gene and (v) a transcription factor (Fig. 1). While the first four search types take genomic regions as input, the transcription factor query performs genome-wide searches, and identifies all variants that alter motif matches for the query transcription factor. Prior to submitting a search request, users can specify P-value thresholds for the changes in motif matches, and motif matches with the SNP and reference alleles. atSNP Search also allows the user to designate multi-level sorting based on the P-values and the genomic coordinates to an output table. Both the user-defined P-value cutoffs and sorting options make the atSNP Search more flexible and convenient compared to existing resources. Further, users may restrict searches to variants that either enhance or disrupt binding, and filter out records based on information contents of the motifs.
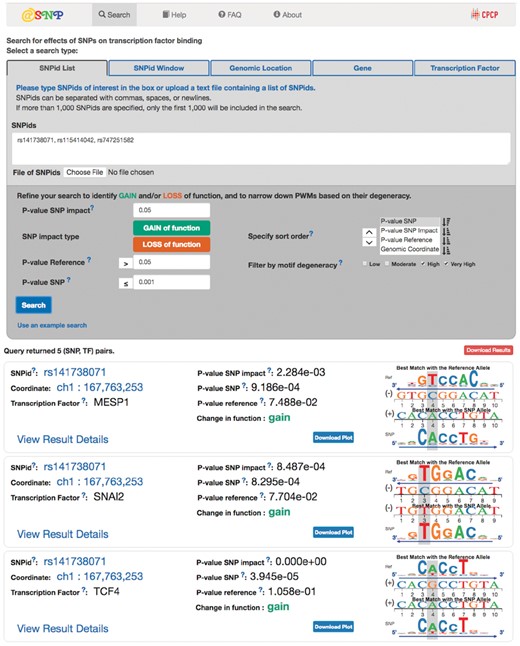
The atSNP Search input form and output table. atSNP Search evaluates both strands of the reference and variant alleles around each SNP location given a motif. The composite sequence logo plot depicts the best matches of both alleles to the motif along with the strand information
The atSNP Search output table provides summary statistics and composite logo plots for variant-motif pairs that meet the search criteria (Fig. 1). Each SNP ID is linked to its dbSNP webpage (Sherry et al., 2001) and the UCSC Genome Browser webpage (Casper et al., 2017) to display the genomic region around the variant position. Users can also access the webpages for transcription factors curated in Factorbook (Wang et al., 2013). Testing results for each variant-motif pair are summarized with three P-values for motif matches with the reference and SNP alleles (P-value Reference, P-value SNP) and the changes in motif matches (P-value SNP Impact) along with the direction of the change. Corresponding composite logo plots display sequence logos aligned to best motif matches with the reference and SNP alleles, and visually reveal the direction of the change in motif match. Composite logo plots are especially useful to validate or reject matches to nearly degenerate motifs. Both the tables and logo plots are downloadable in csv and png formats, respectively. atSNP Search further provides detail pages displaying all statistical results of variant-motif pairs, and hypertext links for JASPAR motifs (Mathelier et al., 2014).
In Supplementary Material, we provide a general purpose use-case for the atSNP Search using variants in the UK Biobank Axiom Array (Allen et al., 2012). This application showcases how such an analysis can identify main transcription factors affected by regulatory SNPs.
Acknowledgements
We are grateful to Matt Ziegler for his guidance on designing the usability test of atSNP Search and Danny Panyard, Kyle Hewitth and Makoto Ohash for participating in the usability test and providing feedback. We also thank the Keleş Research Group for their continuous feedback on atSNP Search.
Funding
This work was supported by National Institutes of Health BD2K grant [U54 AI117924]; National Institutes of Health/National Human Genome Research Institute R01 grant [HG003747]; and U01 grant [HG007019].
Conflict of Interest: none declared.
References


{kind=link}