





Abstract
Combinatorial association mapping aims to assess the statistical association of higher-order interactions of genetic markers with a phenotype of interest. This article presents combinatorial association mapping (CASMAP), a software package that leverages recent advances in significant pattern mining to overcome the statistical and computational challenges that have hindered combinatorial association mapping. CASMAP can be used to perform region-based association studies and to detect higher-order epistatic interactions of genetic variants. Most importantly, unlike other existing significant pattern mining-based tools, CASMAP allows for the correction of categorical covariates such as age or gender, making it suitable for genome-wide association studies.
The R and Python packages can be downloaded from our GitHub repository http://github.com/BorgwardtLab/CASMAP. The R package is also available on CRAN.
Supplementary data are available at Bioinformatics online.
1 Introduction
The goal of genome-wide association studies (GWASs) is to find single-nucleotide polymorphisms (SNPs) that are significantly associated with a trait of interest. However, univariate GWAS often fail to detect associations to rare variants or to variants with weak effects (Burrell et al., 2013), due to the large number of tests performed and the relatively low sample size. Moreover, higher-order epistatic interactions, which may explain part of the phenotypic variation, might be missed by univariate testing. Nevertheless, testing all higher-order interactions between SNPs causes two fundamental difficulties: (i) the number of association tests to perform is too large for naive implementations to be computationally feasible and (ii) the need to correct for multiple hypothesis testing (e.g. Bonferroni correction) creates a substantial decrease in statistical power. Existing association mapping approaches to study epistasis (Cordell, 2002) or to perform region-based association mapping (Lee et al., 2014) alleviate these challenges by reducing the search space to a limited number of combinations of variants chosen a priori (see Supplementary Section S1). However, this selection is generally arbitrary and based on incomplete domain knowledge, with the additional risk that ignoring sets of variants could decrease power.
Recently proposed algorithms (Llinares-López et al., 2015, 2017; Papaxanthos et al., 2016) use state-of-the-art significant pattern mining techniques to overcome those statistical and computational challenges. These algorithms exploit the concept of testability (Tarone, 1990) and implement an efficient pruning criterion in a branch-and-bound fashion (Terada et al., 2013). The combinatorial association mapping (CASMAP) package provides an efficient and user-friendly implementation of these methods. Although the software package allows for general applications of significant pattern mining, it was developed with a strong focus towards GWAS. When compared with Massive Parallel Limitless Arity Multiple-testing Procedure (MP-LAMP) (Yoshizoe et al., 2018), the state-of-the-art significant pattern mining-based software package for GWAS, the contributions of CASMAP are 2-fold: (i) it allows for the correction of covariates, such as age or population structure, which could lead to the detection of spurious associations if not taken into account and (ii) it provides methods to carry out burden tests for genomic regions at any starting position and length, in addition to conducting higher-order epistasis search.
The CASMAP toolbox is easy to install and easy to use. Implemented in C++, it is available both in Python and R and is compatible with both the input format defined by the popular software PLINK (Purcell et al., 2007) and tab-delimited text files. The tool creates output files that contain detailed profiling results, a summary of statistical results and the list of significantly associated regions or sets of genomic variants.
2 Combinatorial association mapping
In this section, we describe the problems that can be tackled by CASMAP. We refer the reader to the Supplementary Material for a detailed description on how to run the tools to address specific use cases.
2.1 Region-based association studies
Testing genomic regions for association is based on the hypothesis that aggregating multiple neighboring SNPs will yield a stronger signal (see Fig. 1). A natural way to perform this type of analysis is to test genomic regions for association with a phenotype of interest. These regions can be predefined, as it is the case in burden tests (e.g. the coding regions of genes). Nevertheless, to avoid restricting ourselves to pre-specified genomic regions, it is preferable to perform a genome-wide analysis of all possible regions. It was shown (Llinares-López et al., 2015) that such an exploration can be efficiently done while retaining statistical power. Additionally, if uncontrolled factors of variation such as age, gender or population stratification are present in the data, a recent algorithm allows for the analysis of all regions while correcting for such covariates (Llinares-López et al., 2017). CASMAP performs region-based association studies, without needing to predefine regions of interest, by integrating into one package the algorithmic properties of the methods mentioned earlier.
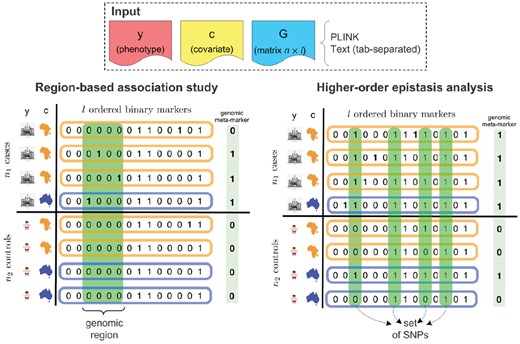
Overview of the two types of combinatorial association mappings supported by CASMAP. The input phenotype y and sample data matrix G are binary. The covariate c is discrete and here represents the genetic ancestry of the individual (correction for population structure). Input is in PLINK format or tab-separated text files. The meta-marker for each individual is created differently depending on the type of analysis: for regions is a Boolean OR and for sets is the Boolean AND (see Supplementary Section S3)
2.2 Higher-order epistasis analysis
Approaches to search for multiplicative interactions of SNPs that are statistically associated with a phenotype (epistasis) predominantly focus on pairwise interactions only. For many of these methods, this limitation is a necessary tradeoff to reduce the number of association tests that would need to be performed. However, by neglecting to consider higher-order interactions, interesting signals might be lost. It was recently shown (Terada et al., 2013) that significant pattern mining techniques can be leveraged to search for significant interactions up to any order. Moreover, recent work (Papaxanthos et al., 2016) extended this algorithm to allow correcting for covariates leading to a method which is applicable to GWAS data with hidden confounders (see Fig. 1). CASMAP provides a GWAS-centric interface to use these two approaches to carry out higher-order epistasis analyses, correcting for covariates if necessary.
3 Features
3.1 I/O files
The input files consist of the sample data, the phenotype and an optional covariate file. After running the analysis, the output of the tools are text files whose contents will depend on which analysis was conducted. For region-based association studies, the main output will consist of significantly associated genomic regions, marked by a start and end positions (SNPs), with their respective P-value. To avoid reporting numerous overlapping regions, a clustering post-processing step is performed and the final output contains the results of this step. In higher-order epistasis analyses, the main output will report the sets of SNPs whose association to the phenotype was found to be statistically significant. Refer to the Supplementary Material for additional details on the contents of these and additional input and output files.
3.2 Correction for covariates
A key feature of CASMAP is its ability to correct for covariate factors in the data (Llinares-López et al., 2017; Papaxanthos et al., 2016). As opposed to other state-of-the-art approaches in significant pattern mining, our tools are well suited to analyze genotype data in the presence of hidden confounders. If the user is interested in performing either type of analysis without correcting for covariates, CASMAP implements two methods that rely on χ2 statistical tests (Llinares-López et al., 2015; Terada et al., 2013).
4 Results
We used CASMAP in a GWAS dataset of near 8000 individuals (Regan et al., 2011). Individuals in the dataset belong to two different subpopulations, thus population structure acts as an important confounder. After binarizing the SNPs according to a dominant encoding, and deriving a covariate from the top six principal components, our tool performed a region-based association study on a total of 615 906 SNPs. It took CASMAP ∼7 min on a single core to identify several associated regions. Furthermore, none of these regions were identified by single SNP association tests or burden tests. Additional details on the biological relevance of the results can be found in Llinares-López et al. (2017).
Funding
This work was supported by the SNSF Starting Grant ‘Significant Pattern Mining’ (to K.B.).
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Felipe Llinares-López and Laetitia Papaxanthos authors should be regarded as Joint First Authors.
The authors wish it to be known that, in their opinion, Dean Bodenham and Karsten Borgwardt authors should be regarded as Joint Last Authors.


{kind=link}