





Abstract
Links are generalization of knots, that consist of several components. They appear in proteins, peptides and other biopolymers with disulfide bonds or ions interactions giving rise to the exceptional stability. Moreover because of this stability such biopolymers are the target of commercial and medical use (including anti-bacterial and insecticidal activity). Therefore, topological characterization of such biopolymers, not only provides explanation of their thermodynamical or mechanical properties, but paves the way to design templates in pharmaceutical applications. However, distinction between links and trivial topology is not an easy task. Here, we present PyLink—a PyMOL plugin suited to identify three types of links and perform comprehensive topological analysis of proteins rich in disulfide or ion bonds. PyLink can scan for the links automatically, or the user may specify their own components, including closed loops with several bridges and ion interactions. This creates the possibility of designing new biopolymers with desired properties.
The PyLink plugin, manual and tutorial videos are available at http://pylink.cent.uw.edu.pl.
1 Introduction
Climbers trust them with their lives, sailors utilize them to stabilize the ships’ sails, and billions of people make them every day to comfortably wear shoes. We use knots and links to stabilize ropes, lines, etc. But the invention of this stabilizing effect cannot be credited to humanity—knots and links appear also in biopolymers such as DNA and proteins providing extra stability to these structures (Dabrowski-Tumanski and Sulkowska, 2017; Zhao et al., 2017).
This extra stability (resistance to harsh conditions or increased mechanical stability) is, however, only one side of the coin. On the other hand, the link topology complicates the dynamics of the polymer chain, e.g. the protein folding. In fact, incorrect order of link formation may lead to protein misfolding (Bronsoms et al., 2011). The situation is even more knotty, as there are at least three ways one can define protein links (Dabrowski-Tumanski et al., 2017). The first are deterministic links, in which the components are formed by the main chain closed by disulfide bridges. The second are probabilistic links in which the components are formed by segments of the main chain and closed by extending the termini toward one distantly located point (‘infinity’). As the closure procedure is performed in a random direction many times (to remove the direction bias), such links are presented with an estimate of their statistical probability. An example of probabilistic links are domain-swapped proteins. The third group is macromolecular links found, for example, in spherical virus capsids. In such links, the components are formed by a few interacting chains. Examples of these types of links are shown in Figure 1.
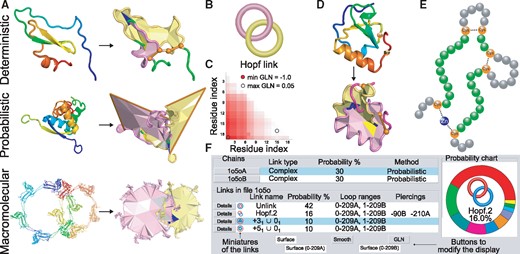
(A) Three types of Hopf link: deterministic, probabilistic and macromolecular. Left column—the original structures (from top to bottom: TdPI, PDB code 2lfk, chain A, arc represor, PDB code 1arr, chains A and B, chains from HK94 virus capsid, PDB code 3j4u), right column—the link visualization in PyLink. For deterministic link the covalent loops are closed by Cys24–Cys51 and Cys52–Cys69 bridges (orange cylinders). For probabilistic link the orange ‘horns’ represent closing the chains by extending them towards ‘infinity’. For convenience, the triangulated surfaces are spanned. (B) The schematic depiction of Hopf Link. (C) Exemplary GLN matrix. (D) Exemplary application of PyLink—adding an artificial bridge between Lys56 and Ser72 (indicated as orange beads, top structure) results in the Hopf link topology, which may be more stable (bottom structure). Such bridge may be possible to create after the mutation Ser->Glu and forming the posttranslational amide bond. (E) The scheme of a closed loop involving ion (Zn2+) and two disulfide bridges. The beads denote residues, the closed loop is marked green. (F) The PyLink result table with different link topologies identified and the pie chart. The table includes the chains involved in the link structure (top part) and the list of links identified (bottom part). For each link, the link miniature, link name, its probability, ranges of the component-forming loops and piercings (if present) are given. Below are the buttons to show/hide the surfaces, to show GLN matrix, or to smooth the structure (Color version of this figure is available at Bioinformatics online.)
This diversity in link types usually makes it impossible to distinguish the link within densely packed structures (e.g. virus capsids). Nevertheless knowing the proteins’ or polymer’s structure topology is indispensable in understand their properties. Moreover, polymers’ links have become an important research target due to their topologically increase stability. This creates the need for an easy tool allowing one to identify, study and design all three types of links in proteins and other polymers. PyLink is an answer to this need.
2 PyLink description and applications
PyLink is a PyMOL plugin designed to identify the deterministic, probabilistic and macromolecular links in proteins. The links can be found both in automatic mode (requiring minimal input from the user) and in manual mode, giving the user the freedom to specify linked components. In the automatic mode, the plugin finds all the necessary data for deterministic and probabilistic links in the PDB file. In the case of macromolecular links, the user needs to enter the names of the chains forming a component (as searching through all chains in the virus capsid would be too time-consuming). Then, PyLink probes all interactions between selected chains and creates the best macrocomponent.
For polymer chains (just the XYZ file), or proteins with no information about inter-chain interactions (e.g. simulation frames), the manual mode can be used. The user may specify the residues, which should be joined directly (the ‘bridge’ for deterministic and macromolecular links), or by connection to one distantly located point (for probabilistic links). Such residues may be entered by their indices or the user can transfer the choice from the sequence or the PyMOL Viewer.
Moreover, PyLink has the unique capability of searching for closed loops formed by a few covalent bridges or interaction via ions (Fig. 1). Slipknotting through such loops is a hallmark of mechanically highly stable proteins (Sikora et al., 2009). Links formed by such loops were already identified (Liang and Mislow, 1994). PyLink can print out all such closed loops (found by searching for cycles in an appropriate graph presentation of the chain), which can then be included in the link analysis.
The links identified are shown in the table, and their probabilities are shown as a pie chart (Fig. 1) (only one link is identified for deterministic and macromolecular links). Moreover the magnitude and direction of windings between loops can be determined based on the Gaussian Linking Number (GLN). Calculating the overall GLN between chains was already used to classify topologically complex structures (Baiesi et al., 2016), however, PyLink calculates also GLN for every subchain of one closed loop against the second closed loop and presents it as a graphical matrix (Fig. 1).
3 Advanced applications
It is well known that cysteine bonds or ion interactions introduce additionally stabilization to structures (Craik et al., 2002), where spatial arrangement gives rise to exceptional stability as in cyclotides or cysteine knots (Cyclotides, 1999). Moreover, these peptides are exceptionally stable to enzymatic degradation. Thus such peptides due to their stability are commonly used as templates in pharmaceutical applications [including anti-HIV, anti-bacterial and insecticidal activity (Craik et al., 2002)].
PyLink is the only tool known to us that performs a comprehensive analysis of biopolymers rich in cysteines bonds and ion interactions. Moreover, it is possible to test the topology by specifying any (also artificial) bridge between residues (see Fig. 1). This capability is crucial for designing new stabilization in proteins or in arbitrary linear polymers, or new templates for organic and synthetic chemistry. Finally, the user may localize the nontriviality in the structure by analyzing the GLN matrices or by probing linkages between different subchains and thus find the key amino acids, e.g. responsible for misfolding in oxidative conditions.
With PyLasso (analyzing lassos) (Gierut et al., 2017), PyKnot (analyzing knots) (Lua, 2012) and cyclotides or cysteine knots described in www.cyclotide.com website one has a set of tools that help to disentangle the proteins’ topological mysteries and to create new, functional, topologically stabilized materials.
4 Technical details
PyLink is written in Python 2.7. It requires PyMOL, Matplotlib and Pillow libraries. The topology is determined with our algorithms and computation of HOMFLY-PT polynomial. Link with up to 4 components can be analyzed simultaneously.
Funding
Support of the National Science Centre [#2016/21/N/NZ1/02848 to PD-T] and The Ministry of Science and Higher Education, Idea Plus grant [#0003/ID3/2016/64 to JIS] is acknowledged.
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Aleksandra M. Gierut and Pawel Dabrowski-Tumanski authors should be regarded as Joint First Authors.


{kind=link}