





Abstract
A better understanding of antibody responses to HIV-1 infection in humans can provide novel insights for the development of an effective HIV-1 vaccine. Neutralization fingerprinting (NFP) is an efficient and accurate algorithm for delineating the epitope specificities found in polyclonal antibody responses to HIV-1 infection. Here, we report the development of NFPws, a web server implementation of the NFP algorithm. The server takes as input serum neutralization data for a set of diverse viral strains, and uses a mathematical model to identify similarities between the serum neutralization pattern and the patterns for known broadly neutralizing monoclonal antibodies (bNAbs), in order to predict the prevalence of bNAb epitope specificities in the given serum. In addition, NFPws also computes and displays a number of estimates related to prediction confidence, as well as the likelihood of presence of novel, previously uncharacterized, antibody specificities in a given serum. NFPws also implements a JSmol viewer for molecular structure visualization of the prediction results. Overall, the NFPws server will be an important tool for the identification and analysis of epitope specificities of bNAb responses against HIV-1.
NFPws is freely available to access at (http://iglab.accre.vanderbilt.edu/NFPws). The webserver is developed using html, CSS, javascript and perl CGI scripts. The NFP algorithm is implemented with scripts written in octave, linux shell and perl. JSmol is implemented to visualize the prediction results on a representative 3D structure of an HIV-1 antigen.
1 Introduction
The envelope glycoprotein (Env) on the surface of HIV-1 facilitates viral entry into host cells by receptor recognition and viral fusion, and is the exclusive target of the neutralizing antibody response (Pancera et al., 2014; Ward and Wilson, 2015; Wyatt and Sodroski, 1998). Isolation and characterization of broadly neutralizing antibodies (bNAbs) from HIV-1 infected individuals directly informs vaccine design efforts (Dosenovic et al., 2015; Georgiev et al., 2013a; Jardine et al., 2015), but the presence of diverse bNAbs targeting various regions of HIV-1 Env makes it difficult to deconvolute epitope specificities present in a given donor’s serum (Bonsignori et al., 2012; Huang et al., 2014; Walker et al., 2010).
Recent computational approaches have accelerated serum epitope mapping by analyzing the serum neutralization data of diverse viral panels (Doria-Rose et al., 2017; Georgiev et al., 2013b; Lacerda et al., 2013; West et al., 2013). In particular, we recently developed and validated a neutralization fingerprinting (NFP) algorithm, which efficiently and accurately predicts epitope-specific antibody responses to HIV-1 infection through computational analysis of the serum pattern of neutralization of a set of diverse viral strains (Doria-Rose et al., 2017; Georgiev et al., 2013b). Briefly, the NFP algorithm compares: (i) the polyclonal serum neutralization data for a given panel of virus strains with (ii) the neutralization patterns for a reference set of known bNAbs over the same viral panel, in order to predict the prevalence of each of the reference bNAb epitope specificities in the given serum. In addition, the algorithm computes a number of measures to quantify prediction accuracy and estimate the likelihood of presence of novel epitope specificities not found in the reference bNAb set. Here, we implemented the NFP algorithm as part of a web server, NFPws. Overall, we expect NFPws to be useful to the HIV-1 immunology community and ultimately aid in the discovery of an effective HIV-1 vaccine.
2 Data input
NFPws accepts serum neutralization values (ID50) of a set of viral strains as input (Fig. 1A) (Georgiev et al., 2013b). All data should be tab-separated, with serum names as the first row and viral strain names as the first column. A detailed description of the input formats and sample files are provided on the website. NFPws optionally requires an input resistance cutoff value for ID50; otherwise, the default value of 40 is assigned. Any serum neutralization data values less than the resistance cutoff are set to that cutoff value before the NFP analysis is applied. Typically, resistance cutoff ID50 values of 40 or 100 could be appropriate, although the user could consider lowering the cutoff for sera with limited neutralization potency. Input data must be for at least one serum and two viral strains. After data format validation, NFPws compares user-provided viral strain names to an internal dataset of viral strains for which monoclonal bNAb data is available on the server, and displays the list of identical and similar strain names (Fig. 1B).
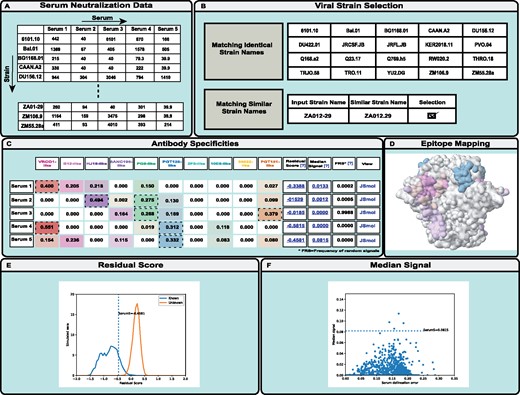
Schematic diagram describing the workflow of the NFPws web server. (A) On the input page, NFPws accepts serum neutralization data in a tab-separated format. (B) On the virus strain selection page, NFPws compares the input data with an internal dataset and displays a list of matching and similar strain names. (C) On the results page, NFPws displays the predicted prevalence values for 10 reference antibody specificities, with additional estimates, such as residual score and median signal. Based on user selection, NFPws also computes the frequency of random signals. (D, E, F) Highlighted visualization options for Serum 5. (D) NFPws provides an option to visualize the predicted antibody specificities for a given serum, by mapping the respective epitopes to a representative antigen structure, on an antibody-specific color spectrum of white (prevalence value = 0) to a set of colors, each of which is specific to one of a reference set of epitope-specific bNAb groups (prevalence value = 1). (E) Distributions of residual scores for simulated sera with known (color: blue) and unknown (color: orange) specificities. The residual score of the input serum (labeled)s is shown as a dotted vertical line. (F) Scatterplot of median scores for the set of simulated sera with known specificities. The median score of the input serum (labeled) is shown as a dotted horizontal line
Data for the identical viral strains are included automatically in the computation. Viral strains with similar but not identical names are highlighted for manual user selection; this is done since there is no single naming nomenclature that is accepted in the field, and the same strain name may have a different spelling in different datasets. In addition to performing the serum epitope delineation, NFPws also computes several estimates of prediction accuracy (Doria-Rose et al., 2017); since one of these estimates, the frequency of random signals, is substantially more computationally intensive than the rest of the algorithm, it is only performed if the user selects the respective checkbox on the input page.
3 Server output and discussion
NFPws compares the user-provided serum neutralization pattern and the patterns for known bNAbs using a mathematical NFP algorithm, in order to predict the prevalence of each of ten reference epitope-specific bNAb groups [CD4-binding site (VRC01-, b12- and HJ16-like), V1/V2 apex (PG9-like), Glycan-V3 (PGT128-like), gp120-gp41 interface (8ANC195-, 35022- and PGT151-like), and membrane-proximal external region (2F5- and 10E8-like)] (Doria-Rose et al., 2017). NFPws displays the prediction results in a tabular format where each row shows the results for a single serum (Fig. 1C). In each row, the first column shows the given serum name; Columns 2–11 show the prevalence of each reference bNAb group; Columns 12–14 provide computed values for the normalized residual score, median delineation score and frequency of random signals, respectively; and the last column provides a JSmol link to visualize the prediction results on a 3D antigen structure.
The prevalence (coefficient) values for the reference bNAb groups range from 0 to 1, and the sum of all 10 values is 1. A coefficient value ≥0.25 is typically considered positive, and higher values denote greater contribution of the respective antibody epitope specificity to serum neutralization. In addition, the computed values for the normalized residual score, median of delineation scores, and frequency of random signals are used to estimate prediction quality for each serum: higher values suggest either a higher serum delineation error, or in the case of a higher residual score—the presence of potentially novel antibody specificities not found in the reference set of bNAbs (Doria-Rose et al., 2017). To assist with the interpretation of these scores, NFPws allows the user to visualize two graphs for each serum. First, a link is provided in the Residual Score column to visualize where the residual score for the input serum falls in comparison to the distributions of the residual scores for simulated sera with known and unknown specificities (Fig. 1E). Second, a link is provided in the Median Signal column to visualize where the median score for the input serum falls in comparison to the median scores for the set of simulated sera with known specificities (Fig. 1F). In addition, the optional Frequency of random signals measure is computed as follows: a set of simulated neutralization fingerprints (that have a low correlation with all bNAbs in the reference set) are added to the reference bNAb set, one at a time, to determine the fraction of fingerprints for which a given input serum has a positive signal (prevalence value for the simulated fingerprint of at least 0.25) (Doria-Rose et al., 2017). For each input serum, NFPws also provides the ability to visualize the antibody specificities by mapping the predicted bNAb prevalence to a representative antigen structure in a JSmol viewer (Fig. 1D). The surface of the antigen is colored based on the same antibody-specific color spectrum as shown in the tabular results, on a scale of white (prevalence value = 0) to full color (prevalence value = 1). Residues that are part of multiple bNAb epitopes are colored according to the epitope with the highest prevalence value for the given serum. The structure used for the epitope/specificity mapping is a structure of HIV-1 Env gp140 (Stewart-Jones et al., 2016).
4 Conclusion
We have developed a web server to implement the NFP algorithm, NFPws, which utilizes serum neutralization data for a set of viral strains to predict the prevalence of epitope-specific bNAb responses. NFPws will allow efficient and accurate delineation of antibody specificities in serum samples from HIV-infected donors.
Acknowledgements
We thank members of the Georgiev laboratory for discussions and comments on the manuscript. This work was conducted in part using the resources of the Advanced Computing Center for Research and Education (ACCRE) at Vanderbilt.
Funding
This work was supported in part by the National Institutes of Health (R01 AI131722: I.S.G., N.R., and I.S.); the Vanderbilt Program for Next Generation Vaccines (N.R.); and the Vanderbilt Molecular Biophysics Training Program (T32GM008320: I.S.).
Conflict of Interest: none declared.
References


{kind=link}