





Abstract
Knowledge graph embeddings (KGEs) have received significant attention in other domains due to their ability to predict links and create dense representations for graphs’ nodes and edges. However, the software ecosystem for their application to bioinformatics remains limited and inaccessible for users without expertise in programing and machine learning. Therefore, we developed BioKEEN (Biological KnowlEdge EmbeddiNgs) and PyKEEN (Python KnowlEdge EmbeddiNgs) to facilitate their easy use through an interactive command line interface. Finally, we present a case study in which we used a novel biological pathway mapping resource to predict links that represent pathway crosstalks and hierarchies.
BioKEEN and PyKEEN are open source Python packages publicly available under the MIT License at https://github.com/SmartDataAnalytics/BioKEEN and https://github.com/SmartDataAnalytics/PyKEEN
Supplementary data are available at Bioinformatics online.
1 Introduction
Knowledge graphs (KGs) are multi-relational, directed graphs in which nodes represent entities and edges represent their relations (Bordes et al. 2013). While they have been successfully applied for question answering, information extraction and named entity disambiguation outside of the biomedical domain, their usage in biomedical applications remains limited (Su et al., 2018).
Because KGs are inherently incomplete and noisy, several methods have been developed for deriving or predicting missing edges (Nickel et al., 2016). One method is to apply reasoning based on formal logic to derive missing edges, but it usually requires a large set of user-defined formulas to achieve generalization. Another method is to train KG embeddings (KGEs; low-dimensional vector/matrix representations of entities and relations whose elements correspond to latent features of the KG) that best preserve the structural characteristics of the KG and then predict new edges using their respective KGE models (Wang et al., 2017).
In a biological setting, relation prediction not only enables researchers to expand their KGs, but also to generate new hypotheses that can be tested experimentally.
Here, we present BioKEEN (Biological KnowlEdge EmbeddiNgs): a Python package for training and evaluating KGEs on biological KGs that is accessible and facile for bioinformaticians without expert knowledge in machine learning through an interactive command line interface (CLI). Through the integration of the Bio2BEL software (https://github.com/bio2bel) within BioKEEN, numerous biomedical databases containing structured knowledge are directly accessible. Additionally, we have externalized BioKEEN’s core machine learning components for training and evaluating KGE models in an independent Python package, PyKEEN, such that they can be reused in other domains (see Fig. 1).
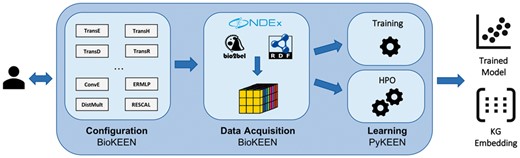
Software architecture of BioKEEN (i) Configuration: Users define experiments through the CLI. (ii) Data Acquisition: Dataset(s) are (down-) loaded and transformed into a tensor. (iii) Learning: The KGE model is trained with user-defined hyper-parameters or a hyper-parameter search is applied to find the best set of hyper-parameter values. The functionality of this layer is externalized in the Python KnowlEdge EmbeddiNgs package
Although there exists other toolkits like OpenKE (Han et al., 2018) and scikit-kge (https://github.com/mnick/scikit-kge), they are not specialized for bioinformatics applications and require more expertise in programming and in KGEs. To the best of our knowledge, BioKEEN is the first framework specifically designed to facilitate the use of KGE models for users in the bioinformatics community.
2 Software architecture
The BioKEEN software package consists of three layers: (i) the model configuration layer, (ii) the data acquisition and transformation layer and (iii) the learning layer (see Fig. 1).
2.1 Configuration layer
Because every KGE model has its own set of hyper-parameters, the configuration of an experiment for a non-expert can be very complicated and discouraging. This possible obstacle is addressed in the configuration layer through an interactive CLI that assists users in setting up their experiments (i.e. defining the datasets, the model, and its parameters). Based on the configuration, BioKEEN builds a machine learning pipeline containing the appropriate components (e.g. data acquisition, training, evaluation and prediction).
Currently, we provide implementations of 10 embedding models (e.g. TransE, TransH, ConvE etc.; Dettmers et al., 2017; Wang et al., 2017). A full list can be found in Supplementary Table S1. Moreover, BioKEEN can be executed in training and hyper-parameter optimization (HPO) mode.
2.2 Data acquisition layer
Because extracting and preparing training data can be a time-consuming process, BioKEEN integrates the Bio2BEL software to download and parse numerous biomedical databases (Supplementary Table S2). This allows users to focus on the experiments, to automatically incorporate the latest database versions, and to have access to new datasets as they are incorporated into Bio2BEL. In addition, users can provide their own datasets as tab-separated values, RDF or from NDEx (Pratt et al., 2015). BioKEEN processes the selected and provided datasets then transforms them into a tensor (i.e. a multi-dimensional matrix) for further processing.
2.3 Learning layer
Determining the appropriate values for the hyper-parameters of a KGE model requires both machine learning and domain specific knowledge. If the user specifies hyper-parameters, BioKEEN can be run directly in training mode. Otherwise, it first runs in HPO mode, where random search is applied to find suitable hyper-parameters values from (user) predefined sets. We implemented random search instead of the widely applied grid search because it converges faster to appropriate hyper-parameter values (Goodfellow et al. 2016). Finally, the user can run BioKEEN in training mode with the resulting hyper-parameter values.
To train the models, negative training examples are generated based on the algorithm described in Bordes et al. To evaluate the trained models, BioKEEN computes two common evaluation metrics for KGE models: mean rank and hits@k.
3 Application
We used BioKEEN to train and evaluate several KGE models on the pathway mappings from ComPath (Domingo-Fernández et al., 2019), the first manually curated intra- and inter-database pathway mapping resource that bridges the representations of similar biological pathways in different databases. Then, we used the best model to predict new relations representing pathway crosstalks and hierarchies. After removing reflexive triplets, we found that the highest ranked novel equivalence between TGF-beta Receptor Signaling (wikipathways: WP560) and TGF-beta signaling pathway (kegg: hsa04350), as well as the highest ranked hierarchical link that Lipoic acid (kegg: hsa00785) is a part of Lipid metabolism (reactome: R-HSA-556833) both represented novel pathway crosstalks. Upon manual evaluation, each fulfilled the ComPath curation criteria and can be added to the resource.
We performed HPO for five different models to illustrate the need for choosing the appropriate hyper-parameter values. For the TransE model, comparing the hyper-parameters similar to those reported by Bordes et al. with the hyper-parameters from HPO showed an improvement in the hits@10 metric from 19.10 to 63.20%.
Moreover, the nature of the model strongly influences the results. We found that the simpler models (e.g. TransE, UM and DistMult) performed similar or even better than the more complex ones (e.g. TransH and TransR). This might be explained by the fact that the more expressive models overfit since ComPath is a not a large dataset. Ultimately, this case scenario illustrates the ability of BioKEEN to assist users in finding reasonable combinations of models and their hyper-parameter values to predict novel links.
4 Discussion and future work
Although BioKEEN already includes several models and components to build machine learning pipelines, it has limitations that could benefit from several additions and improvements.
Modeling multiscale biology (i.e. the -omics, pathway, phenotype and population levels) results in KGs with a variety of compositions, structural features, and topologies for which different KGE models that have not yet been included in BioKEEN may be more appropriate. Further, because of the heterogeneity and lack of structure in most biological and clinical data, we plan to implement additional KGE models that incorporate text, logical rules, and images in addition to the triples in KGs (Hamilton et al. 2018; Wang et al., 2017).
The negative sampling approach described by Bordes et al. included in BioKEEN is prone to false negatives. We plan to mitigate them by incorporating prior biological knowledge and constraints to generate triples guaranteed to be true negatives such as: (i) type constraints for predicates (e.g. the relation transcribed is only valid from gene to protein), (ii) valid attribute range for predicates (e.g. protein weight is below 1000 kDa) and (iii) functional constraints such as mutual exclusion (e.g. a protein is coded by one gene) (Nickel et al., 2016).
Although BioKEEN assists in HPO, it does not provide assistance in selecting a particular KGE model, which is an obscure process even for machine learning experts. We plan to address this by implementing KG analyses with rule-based suggestions (e.g. DistMult performs poorly for KGs with antisymmetric relations).
Finally, we plan to present this software as a web application to enable a wider audience of researchers who may not be comfortable with scripting or CLIs to train and evaluate KGE models.
Acknowledgements
We thank our partners from the Bio2Vec, MLwin and SimpleML projects for their assistance.
Funding
This research was supported by Bio2Vec project (http://bio2vec.net/, CRG6) grant [3454] with funding from King Abdullah University of Science and Technology (KAUST).
Conflict of Interest: none declared.
References


{kind=link}