





Abstract
The performance of docking calculations can be improved by tuning parameters for the system of interest, e.g. biasing the results towards the formation of relevant protein–ligand interactions, such as known ligand pharmacophore or interaction sites derived from cosolvent molecular dynamics. AutoDock Bias is a straightforward and easy to use script-based method that allows the introduction of different types of user-defined biases for fine-tuning AutoDock4 docking calculations.
AutoDock Bias is distributed with MGLTools (since version 1.5.7), and freely available on the web at http://ccsb.scripps.edu/mgltools/ or http://autodockbias.wordpress.com.
Supplementary data are available at Bioinformatics online.
1 Introduction
The AutoDock suite is a widely used free open-source software to perform protein–ligand docking and virtual screening (VS) (Forli et al., 2016; Morris et al., 2009). Recent comparative reviews of popular docking methods showed that success rates are highly system-dependent and performance is similar when the testing set is diverse. Overall results are good for pose prediction, with binding free energy errors of 2–3 kcal/mol for small drug-like molecules in absence of significant receptor conformational adjustment.(Sousa et al., 2013) It is also known that better results can be obtained when the method is adjusted for particular systems using previous knowledge (Cleves and Jain, 2015; Hu and Lill, 2014).
A common way of tuning docking performance is to introduce a restriction or bias towards the formation of a given protein–ligand interaction which is known to be important or essential. For example, in metalloproteins specific ligand groups often coordinate the metal atom. In addition, if several protein–ligand complex structures are available for the same target, a key ligand pharmacophore can be inferred and used to improve docking accuracy (Hu and Lill, 2014). In this context, biased docking has great potential and a wide range of applications.
In the last decade, several strategies have been developed to identify specific protein–ligand interaction sites using small molecular fragments or different solvents. Recently, we showed that water and ethanol sites derived from molecular dynamics (MD) simulations allow to identify over 79% of known protein–ligand interactions, especially those that represent the ligand-derived pharmacophore (Arcon et al., 2017), suggesting that this knowledge could be used to improve docking.
Here, we present AutoDock Bias, a general method that allows guiding AutoDock4 docking calculations towards the formation of any user-defined protein–ligand interaction. As an example, we show how biases from MD-derived solvent sites significantly improve docking both in terms of pose prediction and VS enrichment.
2 Materials and methods
3 Results
The key to AutoDock Bias improved performance is the use of modified maps that enhance AutoDock4 potential driving ligands toward the formation of selected interactions. Figure 1 shows significant improvement, both in terms of pose prediction (Fig. 1A) and VS enrichment (Fig. 1B), after applying biases from solvent sites obtained in water/ethanol MD simulations of AmpC β-lactamase. Two hydrophobic/aromatic and four hydrophilic solvent sites were identified (Supplementary Fig. S1) and used to generate specific biases for appropriate ligand atoms. Results for cross docking of 10 known binders with available crystal structures (Ki below 100 μM, Supplementary Table S1) using standard AutoDock4 and AutoDock Bias are shown in Figure 1A. The plot shows the difference in cluster size (Δpopulation) versus difference in docking score (ΔΔG) between the correctly predicted pose (ligand heavy atom RMSD < 2Å against the reference complex) and the best ranked of the remaining predicted poses. Comparatively, the biased method tends to increase the energy difference and the statistical relevance (i.e. cluster size) between the correct pose and false positive results (upper left quadrant of Fig. 1A). Figure 1B presents comparative ROC curves for a VS application, showing a significantly higher early enrichment with AutoDock Bias (blue solid line) compared with conventional AutoDock4 (green dashed line). Overall, solvent site biased docking improves conventional results for nine targets, with increases up to 0.35 in AUC and up to 7-fold in enrichment factors at 1% (J.P.Arcon et al., 2019, manuscript in preparation.
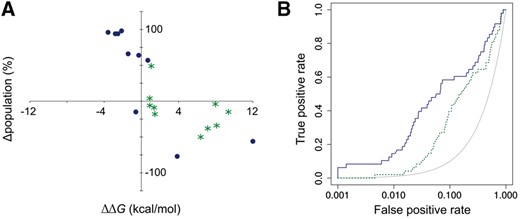
(A) Difference in cluster size (Δpopulation) versus difference in docking score (ΔΔG) between the correctly predicted pose and the best of the remaining predicted poses for cross docking of 10 ligands to AmpC β-lactamase (PDB ID 2r9x). See Supplementary Table S2 for details on each ligand. (B) Semilogarithmic ROC curves for the docking of actives and decoys extracted from the DUD-E database (Mysinger et al., 2012) for AmpC β-lactamase. Results depicted in green (stars, dashed line) for conventional AutoDock4 and blue (dots, solid line) for AutoDock Bias with ethanol solvent sites
Knowledge-based biased docking is widely used and many docking programmes include options to encourage the formation of specific molecular interactions (Corbeil et al., 2012; Friesner et al., 2004; Jones et al., 1997; Ruiz-Carmona et al., 2014) even metal coordination bonds or pose based restraints (tethered docking). In this context, AutoDock Bias complements AutoDock4 by providing a highly versatile, powerful and easy to use tool to improve docking performance, which can be applied in a high-throughput fashion for VS. It allows guided docking towards pharmacophoric interactions (hydrogen bonds, hydrophobic/aromatic) and precise localization of atoms (e.g. metal atoms or anchors for covalent docking) or groups (e.g. substructure core for congeneric ligands or fragment growth) in a defined 3D region relative to the target structure.
Funding
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica [PICT 2015-2276 to A.G.T.], Consejo Nacional de Investigaciones Científicas y Técnicas [PIP 2015-2017 to M.A.M.] and National Institutes of Health [R01 GM069832 to S.F.]. J.P.A. acknowledges Fulbright Commission and Ministerio de Educación de la República Argentina for short term scholarship.
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Juan Pablo Arcon and Carlos P. Modenutti authors should be regarded as Joint First Authors.


{kind=link}