





Abstract
Translational models that utilize omics data generated in in vitro studies to predict the drug efficacy of anti-cancer compounds in patients are highly distinct, which complicates the benchmarking process for new computational approaches. In reaction to this, we introduce the uniFied translatiOnal dRug rESponsE prEdiction platform FORESEE, an open-source R-package. FORESEE not only provides a uniform data format for public cell line and patient datasets, but also establishes a standardized environment for drug response prediction pipelines, incorporating various state-of-the-art pre-processing methods, model training algorithms and validation techniques. The modular implementation of individual elements of the pipeline facilitates a straightforward development of combinatorial models, which can be used to re-evaluate and improve already existing pipelines as well as to develop new ones.
FORESEE is licensed under GNU General Public License v3.0 and available at https://github.com/JRC-COMBINE/FORESEE and https://doi.org/10.17605/OSF.IO/RF6QK, and provides vignettes for documentation and application both online and in the Supplementary Files 2 and 3.
Supplementary data are available at Bioinformatics online.
1 Introduction
Cell line data bases featuring both multi-omics characterizations of human cancer cell lines and their response profiles to drug compounds have become a vital tool in developing predictive drug response models for cancer patients. Concomitant with their advancement, tools have been developed to provide access to the data (Luna et al., 2016; Smirnov et al., 2016) and to systematically evaluate models for drug response prediction (Jang et al., 2014). In order to attain clinical relevance, such cell line-based models need to be translated and tested on patient data, which has become the focus of a steadily growing number of studies: while some are restricted to gene expression data (Geeleher et al., 2014; Huang et al., 2017), other studies additionally incorporate mutation profiles and copy number variations (Dorman et al., 2016), promoter methylation (Aben et al., 2016) and protein expression (Daemen et al., 2013). As a consequence of the diversity and scope of the work that has been performed in this field, comparing the various approaches is complicated and the process of benchmarking novel computational methods has become time-consuming. In order to address this, we introduce the FORESEE platform to facilitate a straightforward and comprehensive evaluation of translational drug response models.
2 Implementation
For the systematic evaluation of individual components of the modeling pipeline and their impact on the performance, the FORESEE package features not only functional elements of the pipeline, but also introduces a common data format for frequently used data resources. Thus, it allows for the methodical investigation of all possible combinations of modeling choices, as well as for testing a specific pipeline on different datasets, thereby exploring how the choice of data affects modeling performance.
2.1 Data
Supporting the idea of translational modeling pipelines, the FORESEE package comprises molecular and pharmacological data that characterize cell lines, xenografts and patients. In terms of cell line characterization, data from the Genomics of Drug Sensitivity in Cancer (Garnett et al., 2012), the Cancer Cell Line Encyclopedia (Barretina et al., 2012; Cancer Cell Line Encyclopedia Consortium and Genomics of Drug Sensitivity in Cancer Consortium, 2015), the Cancer Therapeutics Response Portal (Basu et al., 2013; Rees et al., 2016; Seashore-Ludlow et al., 2015) and Daemen et al. (2013) were formatted into ForeseeCell objects. Each of these ForeseeCell objects contain at least one type of molecular data, such as gene expression, and one type of pharmacological data, such as the IC50 (half maximal inhibitory concentration). On the other hand, information of patients with breast cancer [GSE6434 (Chang et al., 2005) and GSE18864 (Silver et al., 2010)], lung cancer [GSE33072 (Byers et al., 2013)], ovarian cancer [GSE51373 (Koti et al., 2013)] and multiple myeloma [GSE9782 (Mulligan et al., 2007)] was organized into ForeseePatient objects including at least one molecular data type and one measure of in vivo drug efficacy, such as tumor shrinkage. Although primarily developed to design translational models that are trained on cell line data and tested on patient data, FORESEE also allows for testing cell line-trained models on other cell line datasets. Moreover, data from patient derived xenografts (Gao et al., 2015; Witkiewicz et al., 2016), bridging the differences between cell lines and patients, were included as ForeseeCell objects to offer a supplementary translational modeling opportunity: testing patient derived xenografts-trained models on patients. A detailed description of how the data were obtained and prepared can be found in Supplementary File 2.
2.2 Pipeline
The functional elements of the modeling pipeline, which are depicted in Figure 1 and explained in more detail in Supplementary File 1, are implemented as independent modules that can be changed individually, according to the user’s preferences. Across all main steps of the pipeline, user-defined functions can substitute the pre-implemented methods to enable a more flexible use of the package, which is explained in Supplementary File 3 along with other use cases.
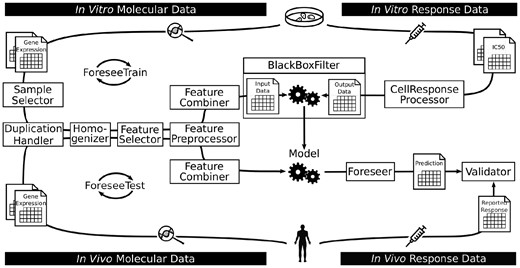
Illustration of the general FORESEE pipeline. The modeling routine comprises two main shells, ForeseeTrain (upper loop) and ForeseeTest (lower loop), with each consisting of different functional elements (boxes). During training, molecular cell line data are pre-processed by selecting certain samples in SampleSelector, removing duplicated feature names in DuplicationHandler, reducing batch effects in Homogenizer, selecting certain features in FeatureSelector, transforming the data in FeaturePreprocessor and combining the different molecular data types in FeatureCombiner, while the response data are transformed in CellResponseProcessor. The pre-processed data are then used for model training in BlackBoxFilter. The Foreseer applies the completed model to molecular patient data that have been pre-processed in the same manner as the cell line data to yield a prediction for patient drug sensitivity, which is subsequently compared to the actual response in Validator to evaluate the overall performance of the translational model
3 Discussion
The FORESEE R-package is designed to explore and compare translational drug response models. Thus, it comprises both a standardized data format for molecular in vitro and in vivo data and functional building blocks that summarize various well-established pre-processing and processing options. Moreover, each of the functional blocks allows for the application of user-defined alternatives to support the fast and easy development of novel modeling pipelines. Future expansions of FORESEE are directed toward an automatic optimization for identifying the modeling pipeline best-suited for a particular setting. Until then, we hope that FORESEE can facilitate exchanging expertise among researchers by providing a standard environment for translational drug sensitivity models and therefore push forward the potential to predict drug sensitivity of cancer patients.
Acknowledgements
We thank Jérôme Schätzle for his assistance in creating the figure and testing the package, and Pejman Farhadi for proof-reading the manuscript.
Funding
This work was partially supported by Bayer AG.
Conflict of Interest: Andreas Schuppert holds a minor, part-time position at Bayer AG. Bayer AG had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
Author notes
The authors wish it to be known that, in their opinion, Lisa-Katrin Turnhoff and Ali Hadizadeh Esfahani authors should be regarded as Joint First Authors.


{kind=link}