





Abstract
We developed the metagenomeFeatures R Bioconductor package along with annotation packages for three 16S rRNA databases (Greengenes, RDP and SILVA) to facilitate working with 16S rRNA databases and marker-gene survey feature data. The metagenomeFeatures package defines two classes, MgDb for working with 16S rRNA sequence databases, and mgFeatures for marker-gene survey feature data. The associated annotation packages provide a consistent interface to the different databases facilitating database comparison and exploration. The mgFeatures-class represents a crucial step in the development of a common data structure for working with 16S marker-gene survey data in R.
https://bioconductor.org/packages/release/bioc/html/metagenomeFeatures.html.
Supplementary material is available at Bioinformatics online.
1 Introduction
16S rRNA marker-gene surveys have significantly advanced our understanding of the diversity and structure of prokaryotic communities in ecosystems including the human gut, open ocean and even the international space station. These surveys use targeted assays to sequence the 16S rRNA gene. The raw sequence data is processed using a bioinformatic pipeline where the sequences are grouped into features, e.g. operational taxonomic units or sequence variants, yielding a set of representative sequences (Fig. 1).
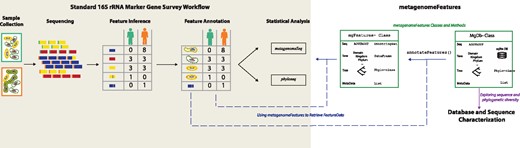
Role of metagenomeFeatures in a 16S rRNA marker-gene survey association study workflow. Standard workflow indicated with the shaded region on the left including sample collection, sequencing, feature inference, feature annotation, then statistical analysis e.g. differential abundance testing and diversity analysis. The R/Bioconductor packages metagenomeSeq and phyloseq are two main utilities for statistical analysis. Contributions by metagenomeFeatures and associated database packages to the standard workflow depicted on right. Arrow and box color indicate metagenomeFeatures vignettes demonstrating functionality. Dashed arrows are connections between standard workflow and metagenomeFeatures
A critical step in 16S rRNA marker-gene survey workflow is feature annotation, comparing representative sequences to a reference database for taxonomic classification or phylogenetic placement (Fig. 1). There are numerous 16S rRNA reference databases of which Greengenes, RDP and SILVA are widely used (Cole et al., 2014; McDonald et al., 2012; Quast et al., 2013). Additionally, there are smaller system-specific databases such as the soil reference database (Choi et al., 2017). 16S rRNA databases differ in the number and diversity of sequences, the taxonomic classification system and the inclusion of intermediate ranks (Balvočiūtė and Huson, 2017). These separate databases present a significant barrier to performing the same analysis using multiple databases since: (i) the data are formatted differently, and sequence identifiers are unique to each database challenging membership and composition comparisons, and (ii) taxonomic assignments can be database-dependent (Pettengill and Rand, 2017). To facilitate database comparisons RNACentral (http://rnacentral.org/), a resource combining non-coding RNA databases, provides unique identifiers for the sequences (The RNAcentral Consortium, 2017).
The R programing language provides a rich environment and software for data analysis (R Core Team, n.d., https://www.R-project.org). Additionally, Bioconductor, the R bioinformatic software resource (Huber et al., 2015) includes packages for working with DNA-sequence data and 16S rRNA marker-gene survey data. Although a number of software tools are available for working with 16S rRNA marker-gene survey feature data, there are no tools for working with multiple 16S rRNA databases. Furthermore, tools for working with 16S rRNA marker-gene survey feature data in R use different data structures. Therefore, an R package defining consistent data structures for working with multiple 16S rRNA databases and marker-gene survey feature data are needed.
To address this need we developed the R package metagenomeFeatures. metagenomeFeatures provides a common data structure for working with the 16S rRNA databases and marker-gene survey feature data. In practice, marker-gene survey feature annotations can be updated or changed easily with new reference databases. For example, the reference phylogenetic tree from one database can be used when a different database was used to initially annotate marker-gene survey features. The RDP database does not include a reference phylogenetic tree but the Greengenes and SILVA database do. One could use the RNAcentral IDs for a marker-gene survey dataset’s RDP taxonomic annotations to annotate the dataset with the Greengenes or SILVA reference phylogenetic tree (Fig. 1). This package is the first step towards developing a common data structure for analyzing metagenomic and marker-gene survey data using R packages.
2 metagenomeFeatures package
The metagenomeFeatures package defines two data structures, MgDb for working with 16S rRNA databases, and mgFeatures for marker-gene survey feature data. There are three types of relevant information for both MgDb and mgFeatures-class objects, (i) the sequences themselves, (ii) sequence taxonomic lineage, and (iii) a phylogenetic tree representing the evolutionary relationship between features.
MgDb and mgFeatures data structures are both S4 object-oriented classes with slots for taxonomy, sequences, phylogenetic tree and metadata. The MgDb-class provides a consistent data structure for working with different 16S rRNA databases. 16S rRNA databases contain hundreds of thousands to millions of sequences. Therefore, an SQLite database is used to store the taxonomic and sequence data. Using an SQLite database prevents the user from loading the full database into memory. We developed Bioconductor annotation packages for the Greengenes, RDP and SILVA databases. Along with database specific sequence identifiers, RNAcentral identifiers are included in the SQLite table for inter-database comparisons. The mgFeatures-class is used for storing and working with marker-gene survey feature data. As the number of features in a marker-gene survey dataset is significantly smaller than the number of sequences in a reference database, mgFeatures-class uses Bioconductor data structures instead of an SQLite database.
The metagenomeFeatures package includes three vignettes with example use cases (Fig. 1). For a list of package vignettes use the R command browseVignettes(‘metagenomeFeatures’). Individual vignettes are viewed using the vignette(‘x’) command replacing x with the name of the vignette you are interested in using. The Supplementary Material characterizes the overlap between the three databases and demonstrates using metagenomeFeatures and the annotation packages to evaluate the potential for species-level taxonomic classification using 16S rRNA sequence data.
3 Conclusions
The metagenomeFeatures package provides data structures and functions for working with 16S rRNA gene sequence reference databases and marker-gene survey feature data. The data structure provided by the MgDb-class in conjunction with the shared sequence identifier system developed by RNACentral facilitates comparisons between 16S rRNA databases. The mgFeatures-class provides the groundwork for the development of a common data structure for working with metagenomic and marker-gene sequence data in R. Additionally, while the data structures were developed for 16S rRNA gene sequence data they can be used for any marker-gene sequence data without modification and can be extended to work with shotgun metagenomic sequence data and databases.
Acknowledgements
The authors would like to thank Dr. Mihai Pop, Dr. Marc Salit, Dr. Samuel Forry and Dr. Arlin Stolzfus for feedback on the article. The Bioconductor core team provided valuable feedback during the package submission and update process. Opinions expressed in this paper are the authors’ and do not necessarily reflect the policies and views of NIST or affiliated venues. Certain commercial equipment, instruments or materials are identified in this article in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendations or endorsement by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose. Official contribution of NIST; not subject to copyrights in USA.
Funding
This work was partially supported by National Institutes of Health (NIH) [NIH RO1GM114267 to J.W., J.K., H.C.B. and NIH R01HG005220 to H.C.B.].
Conflict of Interest: none declared.
References
R Core Team. (n.d.)


{kind=link}