





Abstract
Piwi-interacting RNAs (piRNAs) are a class of small non-coding RNAs which guide endonucleases to mRNAs of actively transcribed transposons in order to prevent their translation. The resulting mRNA fragments induce a positive feedback loop (the ‘ping-pong cycle’), which reinforces piRNA production and hence the transposon-silencing effect. PingPongPro is a command-line tool to scan small RNA-Seq data for signs of ping-pong cycle activity. It implements a novel algorithm that combines empirical probabilities in a multi-factor model to accurately identify transposons which are suppressed through the ping-pong cycle.
Source code, a user manual, and binaries for Microsoft Windows and Linux are available at https://github.com/suhrig/pingpongpro under the GPLv3 license.
Supplementary data are available at Bioinformatics online.
1 Introduction
The destructive activity of transposons is suppressed by Piwi-interacting RNAs (piRNAs), a class of small non-coding RNAs, predominantly active in the germline. They form complexes with Piwi-like endonucleases and guide them to mRNA of actively transcribed transposons by means of complementary base-pairing. The endonucleases degrade the mRNA molecules and thus defuse their harmful potential. The fragments of the degraded mRNA induce the production of even more piRNAs, thus accelerating the degradation of transposon mRNA. This feed-forward loop has been suggested by Brennecke et al. (2007) and Gunawardane et al. (2007), who coined the term ‘ping-pong cycle’ for the process.
Next-generation sequencing (NGS) of small RNA molecules gives insight into ping-pong cycle activity and the transposons suppressed by it. The cycle produces a characteristic pattern of how reads align to the reference genome: since the very nature of the cycle is to multiply piRNA molecules, an active cycle manifests as many reads mapping to the same start coordinate forming a ‘stack’ of reads (Supplementary Fig. S1). Typically two stacks align to opposite strands—one represents fragments of transposon mRNA; the other represents their complementary piRNAs. The endonucleases involved in the ping-pong cycle cleave RNA molecules with an offset of 10 nt, such that the stacks overlap by the first 10 nt from the 5' end. Such pairs of stacks (‘ping-pong signatures’) are hallmarks of piRNA-mediated transposon-silencing and form the basis for the analysis of small RNA-Seq data for signs of ping-pong cycle activity.
2 Algorithm
Naïve algorithms for the detection of ping-pong cycle activity rely solely on read counts (Antoniewski, 2014; Han et al., 2015; Li et al., 2009). They count the number of reads within the region of a transposon overlapping by 10 nt and compare this number against the amount of reads which overlap by arbitrary lengths. If the read count of the former is significantly higher, the transposon is assumed to be suppressed by the ping-pong cycle. Statistical significance is assessed by means of a Z-test.
PingPongPro employs a similar algorithm (Supplementary Fig. S2), but instead of using raw read counts, it uses weighted counts. Each read is assigned a weight equal to the empirical probability that the read is ping-pong-derived. The probabilities are estimated based on how well the read satisfies the characteristics of a true ping-pong signature:
Reads of true ping-pong signatures form stacks, which are aligned at their 5' ends and which overlap with a stack on the opposite strand by exactly 10 nt.
The stack is considerably higher than other stacks in the close vicinity, such that the height cannot be explained by the local coverage.
Often, one of the stacks has adenine at the 10th position.
Supplementary Table S2 and Figure S5 demonstrate the superiority of PingPongPro over the naïve algorithm. On average PingPongPro detects 13% more regions with ping-pong cycle activity without compromising specificity and increases the area under the curve of the receiver-operating characteristic by 0.12.
3 Implementation
PingPongPro was written in C++ and builds on the NGS analysis library SeqAn (Döring et al., 2008). Source code and readily usable binaries are available for Microsoft Windows and Linux.
Accepted input formats for sequencing data are the SAM and BAM file formats. The tool can be supplied with a list of annotated transposons in various formats (GFF, GTF, BED, CSV, TSV). These transposons are checked for ping-pong cycle activity.
PingPongPro outputs all of its results in tab-separated text files, which are easily imported into common statistics/spreadsheet programs. Moreover, it generates bedGraph files for visualization of detected ping-pong signatures and suppressed transposons in popular genome browsers (Fig. 1).
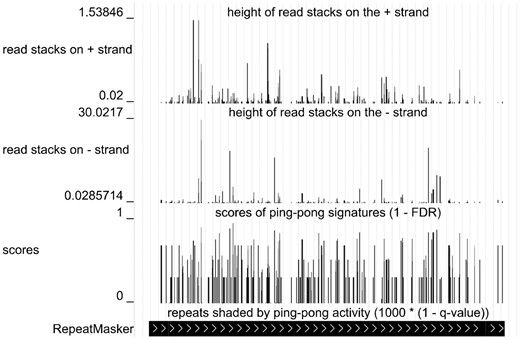
Screenshot of the UCSC genome browser showing the transposon BLOOD_I-int of Drosophila melanogaster (chr2L: 1220184-1227592) covered with ping-pong signatures (data from Li et al., 2009, SRA accession number SRR010960)
4 Conclusion
PingPongPro is a software tool to detect ping-pong cycle activity in small RNA-Seq data. It locates sites of piRNA-mediated cleavage and identifies transposons suppressed through the ping-pong cycle. PingPongPro’s algorithm enhances established methods for the detection of ping-pong cycle activity. By taking additional covariates into account and auto-tuning its parameters to the data, it achieves more reliable predictions than previous approaches. Cross-platform compatibility, support of standard input file formats, flexible output file formats, and a resource-efficient implementation enable researchers to adopt the software with ease.
Acknowledgements
We thank the bioinformatics core facility and the research group of René Ketting at the Institute of Molecular Biology Mainz for their input and data.
Conflict of Interest: none declared.
References


{kind=link}