





Abstract
Since the idea of pan-genomics emerged several tools and pipelines have been introduced for prokaryotic pan-genomics. However, not a single comprehensive pipeline has been reported which could overcome multiple challenges associated with eukaryotic pan-genomics. To aid the eukaryotic pan-genomic studies, here we present ppsPCP pipeline which is designed for eukaryotes especially for plants. It is capable of scanning presence/absence variants (PAVs) and constructing a fully annotated pan-genome. We believe with these unique features of PAV scanning and building a pan-genome together with its annotation, ppsPCP will be useful for plant pan-genomic studies and aid researchers to study genetic/phenotypic variations and genomic diversity.
The ppsPCP is freely available at github DOI: https://doi.org/10.5281/zenodo.2567390 and webpage http://cbi.hzau.edu.cn/ppsPCP/.
Supplementary data are available at Bioinformatics online.
1 Introduction
The recent advancement in plant genomics research has been rapidly developed which results in large volume of data submission in public databases and enable data acquisition, whole-genome sequence data analyses and comparative genomic analyses through different pipelines (Veras et al., 2018). The intra-species genetic variations, especially in the form of presence/absence variants (PAVs) are a key to natural and artificial selection (Gordon et al., 2017). Whole-genome re-sequencing data is usually mapped to a reference genome when genomes of different individuals are compared. However, one single reference genome is insufficient to epitomize genetic makeup of different individuals in the same species and often ignore some important genes and leads to inaccurate estimation of genetic diversity (Schatz et al., 2014). To get a comprehensive map of genetic variations, phenotypic variations and genomic diversity, it is crucial to construct a pan-genome including all the specific PAVs (Giordano et al., 2018).
In this note, we present ppsPCP, a novel pipeline which takes advantage of assembled plant genomes, screen PAVs from them and develops a completely annotated pan-genome.
2 Materials and methods
An overview of the ppsPCP pipeline workflow is shown in Figure 1. When comparing one or multiple query genomes to the reference genome, ppsPCP scans sequences present in query genome but absent in the reference genome, filters genes associated with PAVs and constructs a fully annotated pan-genome. The basic steps of ppsPCP pipeline are as follows: (i) Whole-genome comparison is performed to find out the sequences present in query genome but absent in the reference genome. Whole-genome alignment helps to filter not only genes but also non-coding regions. MUMmer (Kurtz et al., 2004) is used for alignment at this step: first NUCmer utility calculates the delta file and then show-coords package analyzes the output; (ii) The output alignment is parsed to scan PAVs with the default minimum PAV length set to 100 bp; (iii) To appraise and confirm the presence of PAV regions, BLASTn (Camacho et al., 2009) is performed between reference and query genome; (iv) The output of BLASTn is parsed to classify PAVs into two categories: the first category contains sequences highly similar to the reference genome. Similarity 95% and coverage 90% are set as default parameters. All the scanned sequences higher than the criteria are eliminated. The second category contains the sequences which are not found in the reference genome; (v) Filtered PAVs are compared with the query genome, overlapped PAVs are extended and their boundaries are corrected, so they can cover the whole genomic regions (Supplementary Fig. S1). These extended PAVs are used for downstream analysis; (vi) Scanned PAVs are labeled and merged into a single sequence. 100 bp sequence chunks of N-bases are added after every PAV sequence to keep them unique and separate. Annotation file for these scanned PAVs is prepared by harvesting the overlapped genes from their query genome annotation file; (vii) A sequence based draft pan-genome is prepared by merging the scanned PAVs file and its annotation with the reference genome; (viii) Query genome is aligned against the draft pan-genome using BLAT (Kent, 2002) to extract genes not present in the draft pan-genome or at-least not fulfilling one of the previous defined criteria of PAVs scanning. Similarity 80% is set as default parameter to filter mapped genes; (ix) The extracted genes’ regions are mined from query genome and merged with the output files of PAVs yielded at step (v) after boundaries correction. This file is further used for final process; (x) Merged file is compared with the query genome. Regions overlapped with each other are combined, shorter regions are extended, and boundaries are corrected. Sequences of corrected regions are harvested from query genome. All sequences are labeled with the query name and merged into a single sequence file. 100 bp sequence chunks of N-bases are added after each sequence to keep them unique and separate. Annotation file is prepared by harvesting the overlapped genes from their query genome annotation. Finally, a comprehensive sequence and gene-based pan-genome is constructed by merging the scanned sequence file and its annotation with the reference genome.
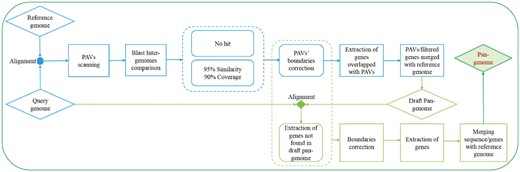
Overview of the ppsPCP pipeline
When running ppsPCP, some options are available, and users can set parameters according to their requirements (details in Supplementary Material Note S1). To run ppsPCP, some additional tools are required to be pre-installed on user’s system. List and details of dependencies are given in Supplementary Material Note S2. Here, ppsPCP is benchmarked with model cereal species rice and model dicot species Arabidopsis thaliana.
3 Results and discussion
ppsPCP is written in Perl programing language and Shell scripting. It is currently available for Linux based platforms. To scan three rice sized genomes (∼400 MB each) and make their pan-genome, ppsPCP only takes about 25 CPU hours. All the useful output information is written in simple txt or log files. ppsPCP accepts genome information in fasta (all extensions like *.fa, *.faa or *.fasta are accepted) and *.gff3/*.gff formats, and outputs pan-genome in fasta/gff3 files. Along with ppsPCP package, a user manual including details about the required software and libraries, explanation for each parameter, guide lines about each available user option and a step-by-step demonstration is given at https://doi.org/10.5281/zenodo.2567390 and http://cbi.hzau.edu.cn/ppsPCP/. In case of rice (details in Supplementary Material Note S3), ppsPCP constructed a 420 MB sized pan-genome containing 43 082 genes. A total of 11 677 PAVs and 4213 genes were screened and added to the rice pan-genome. In case of A.thaliana (details in Supplementary Material Note S3), ppsPCP constructed a 122 MB sized pan-genome containing 34 899 genes. A total of 7480 PAVs and 1432 genes were screened and added to the A.thaliana pan-genome. All the input and output data can be downloaded from ppsPCP webpage. Furthermore, to evaluate the quality of developed pan genomes by ppsPCP, we compared our rice results with recently reported pan-genome developed from 3010 diverse accessions of Asian cultivated rice (Wang et al., 2018) and A.thaliana results with pan-genome of 19 ecotypes (Gan et al., 2011), which is analyzed by using GET_HOMOLOGUES-EST pipeline (Contreras-Moreira et al., 2017). GET_HOMOLOGUES-EST only performs customizable plant pan-genome analysis and cluster orthologous genes using multiple algorithms. In case of rice, only 2650 genes out of 50 955 genes were found to be different, and 48 305 genes were fully mapped to ppsPCP constructed pan-genome. Although in case of A.thaliana, the constructed pan-genome of ppsPCP contains 1687 more genes compared with the pan-genome analyzed by GET_HOMOLOGUES-EST pipeline (which comprises 33 212 genes). These results showed the high accuracy, efficiency and utility of ppsPCP.
4 Conclusion
The prodigious genomic diversity of a genus is a key to identify unique genes for the improvement of its cultivated species. However, one reference genome is inadequate to represent all the gene pool of a species. Advancements in pan genomics are required to better understand functional significance of differentially present genes in eukaryotes. With ppsPCP, it is easy to scan PAVs/genes from different genomes and develop a fully annotated pan-genome. The ppcPCP pipeline will be useful to study genetic/phenotypic variations and their links with useful traits, and genomic diversity within a eukaryotic species.
Funding
This work was supported by the National Key Research and Development Program of China [2016YFD0100904 and 2018YFD1000101], the National Natural Science Foundation of China [31871269 and 31571351] and the Fundamental Research Funds for the Central Universities [2662017PY043].
Conflict of Interest: none declared.
References


{kind=link}