





Abstract
In silico prediction tools are essential for identifying variants which create or disrupt cis-splicing motifs. However, there are limited options for genome-scale discovery of splice-altering variants.
We have developed Spliceogen, a highly scalable pipeline integrating predictions from some of the individually best performing models for splice motif prediction: MaxEntScan, GeneSplicer, ESRseq and Branchpointer.
Spliceogen is available as a command line tool which accepts VCF/BED inputs and handles both single nucleotide variants (SNVs) and indels (https://github.com/VCCRI/Spliceogen). SNV databases with prediction scores are also available, covering all possible SNVs at all genomic positions within all Gencode-annotated multi-exon transcripts.
Supplementary data are available at Bioinformatics online.
1 Introduction
Splicing defects occur in approximately one-third of disease-associated genetic variants (Lim et al., 2011). Variants may alter splicing by directly impacting trans-acting splicing factors or more commonly, by creating or disrupting instances of the cis-acting motifs which guide splice site definition: donors, acceptors, branchpoints, enhancers and silencers. These motifs, which are bound by components of the spliceosome and other splicing factors, exhibit substantial heterogeneity. Many prediction algorithms are available which provide scores that reflect the strength of a motif, or confidence that a motif will be bound in vivo. The American College of Medical Genetics and Genomics guidelines for the interpretation of splicing variants recommends employing multiple prediction algorithms to account for their individual strengths and biases (Richards et al., 2015). Several web and graphical interfaces provide multi-algorithm consensus predictions for influencing any of the cis motifs of splicing, and dbscSNV (Jian et al., 2014) provides a database of ensemble predictions for single nucleotide variant (SNVs) within splice sites. However, the options for comprehensive, genome-scale assessment of variant spliceogenicity are limited. We have developed Spliceogen, a highly scalable tool for the discovery of splice-altering variants which integrates predictions from MaxEntScan (Yeo and Burge, 2004), GeneSplicer (Pertea et al., 2001), ESRseq (Ke et al., 2011) and Branchpointer (Signal et al., 2018).
2 Methods and results
2.1 Algorithm integration and adaptation
Spliceogen is a command line tool that accepts VCF/BED inputs and provides motif scores for both SNVs and indels calculated by multiple prediction algorithms (detailed workflow is shown in Supplementary Fig. S1). The algorithms we selected for Spliceogen cover all the major cis motifs which guide splicing (Fig. 1A). In order to integrate these algorithms, it was necessary to develop several modifications and extensions to the command line implementations of MaxEntScan and GeneSplicer to allow their use in batch variant analysis. First, neither MaxEntScan nor GeneSplicer outputs variant information alongside their predictions or allows for direct comparison between reference and alternative allele scores. We implemented solutions to these issues, e.g. by modifying GeneSplicer to read variant information from a FASTA header and output it alongside predictions. Second, since a variant can occur within any position of a motif, it is necessary to scan the sequence flanking a variant to identify potential motifs. MaxEntScan lacks this functionality, requiring a 9 or 23 bp input string aligned with the respective motifs a priori. We developed scanning functionality for MaxEntScan and ESRseq (detailed in Supplementary Fig. S2), which is similar to the sliding window algorithm recently implemented for a MaxEntScan Ensembl variant effect predictor plugin (Shamsani et al., 2018). Third, GeneSplicer scans the input sequence as well as the reverse complement, regardless of transcript orientation. We modified GeneSplicer to read strand information and restrict scanning only to the given orientation. Comparing the predictions of the original and adapted versions of GeneSplicer in a variant analysis revealed that 26% (12/45) of the original top candidate variants were false positives arising from donor/acceptor-like sequences present on the non-coding strand. Additionally, GeneSplicer reads only one FASTA line per file, substantially limiting its scalability. We adapted GeneSplicer to handle large input files, enabling a 50-fold speed improvement (Supplementary Table S1).
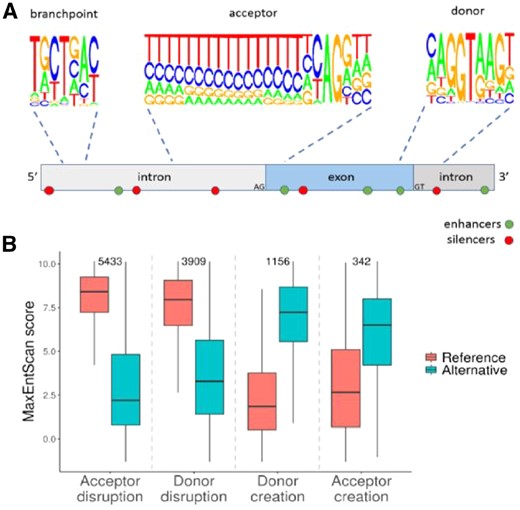
(A) Splicing is guided by donor, acceptor, branchpoint, enhancer and silencer motifs. The motif logo plots were created by deriving the nucleotide frequencies for donor and acceptor motifs from 391 464 internal exon junctions from Gencode-annotated transcripts. Branchpoint motif nucleotide frequencies were derived from 8759 human branchpoints annotated as ‘canonical’ (Taggart et al., 2017). Enhancer and silencer motifs are dispersed throughout. (B) MaxEntScan reference and alternative scores for donor/acceptor creating and disrupting variants. Sample sizes are indicated above. Variants which create new motifs within existing splice sites were excluded
2.2 Identification of splice-altering variants
We applied Spliceogen to a set of 14 438 reported cancer-associated splice-altering variants (Shiraishi et al., 2018). We investigated the reference and alternative MaxEntScan scores separately for donor/acceptor creating and disrupting variants (Fig. 1B). In order to identify potential donor/acceptor disrupting variants, we developed an annotation-based approach for identifying all variants that overlap the extended (9 and 23 bp) donor and acceptor motifs of splice sites, based on the user-provided GTF.
To aid in variant interpretation, we provide ranked candidate variants for different modes of splice disruption. In order to refine our classification of donor/acceptor creating variants, we developed a logistic regression model (Supplementary Material) based on the MaxEntScan and GeneSplicer scores of Shiraishi et al. variants, using random selections of variants outside of splice sites from 1000 Genomes Project (Auton et al., 2015) as a negative dataset. We achieved area under the curve values of 0.952 (donor) and 0.914 (acceptor). Variants are assigned a probability value reflecting their potential to create donor or acceptor splice sites. Supplementary Figure S3 further details our approach for identifying variants which create or disrupt acceptors, donors, branchpoints, enhancers and silencers.
2.3 Scalability and database
Benchmarking was performed on a single compute node with 1 CPU allocated using multiple VCF inputs containing up to 25 million variants. Predictions were generated at a rate of 2.3 million variants per compute hour, with peak memory usage <500 MB (Supplementary Table S2). Benchmarking was performed without including Branchpointer predictions, as it required no adaptation for batch variant analysis.
We used Spliceogen to assess 4.9 billion SNVs, covering all exonic and intronic genomic positions. By selecting all SNVs that either overlap an annotated splice site or receive a high logistic regression score, we provide a comprehensive database of predictions for SNVs with the potential to alter splicing via the creation or disruption of donor/acceptor motifs. In contrast, the coverage of dbscSNV is restricted to 5 million positions adjacent to splice sites.
3 Conclusion
Spliceogen is an integrative, all-in-one pipeline for comprehensive discovery of variants with the potential to alter splicing by creating or disrupting splicing cis motifs. It is available as a highly scalable command line tool as well as a genome-wide SNV database suitable for ANNOVAR annotation (Wang et al., 2010).
Funding
This work was supported by the Chain Reaction (The Ultimate Corporate Bike Challenge to S.L.D.), the Office of Health and Medical Research, NSW State Government to S.L.D., the National Health and Medical Research Council Principal Research Fellowship [1135886 to S.L.D.], the NSW Health Early-Mid Career Fellowship to E.G. and the National Heart Foundation of Australia Future Leader Fellowship [101204 to E.G.].
Conflict of Interest: none declared.
References


{kind=link}