





Abstract
Detection of maximal exact matches (MEMs) between two long sequences is a fundamental problem in pairwise reference-query genome comparisons. To efficiently compare larger and larger genomes, reducing the number of indexed k-mers as well as the number of query k-mers has been adopted as a mainstream approach which saves the computational resources by avoiding a significant number of unnecessary matches.
Under this framework, we proposed a new method to detect all MEMs from a pair of genomes. The method first performs a fixed sampling of k-mers on the query sequence, and adds these selected k-mers to a Bloom filter. Then all the k-mers of the reference sequence are tested by the Bloom filter. If a k-mer passes the test, it is inserted into a hash table for indexing. Compared with the existing methods, much less number of query k-mers are generated and much less k-mers are inserted into the index to avoid unnecessary matches, leading to an efficient matching process and memory usage savings. Experiments on large genomes demonstrate that our method is at least 1.8 times faster than the best of the existing algorithms. This performance is mainly attributed to the key novelty of our method that the fixed k-mer sampling must be conducted on the query sequence and the index k-mers are filtered from the reference sequence via a Bloom filter.
Supplementary data are available at Bioinformatics online.
1 Introduction
Let R be the reference sequence in a genome-to-genome comparison and Q be the query sequence. Suppose subR be a substring from R and subQ be a substring from Q, then subR and subQ are maximal exact matches (MEMs) between R and Q if they are identical and non-extendable to the left or right-side of R or Q (i.e. with a maximum length). MEMs have been widely used in whole-genome alignment (Kurtz et al., 2004), reads alignment (Liu and Schmidt, 2012), long reads error correction (Miclotte et al., 2016) and referential genome compression (Liu et al., 2017). In fact, the detection problem is to find all MEMs from R and Q that have a length longer than a threshold.
MEMs detection has been addressed with two steps by the existing methods: (i) build an index of string patterns from the reference sequence R; (ii) search-and-extend matches using the query sequence Q. The second step usually covers three procedures: (1) search short matches on the index; (2) extend to maximal matches and (3) remove duplicate matches. The efficiency of the second step is heavily affected by the size of the index structure generated from step (i). Many advanced indexing algorithms have been proposed for improving the index construction. For example, MUMmer (Kurtz et al., 2004) build a suffix tree index structure from the reference genome. Although the construction is in linear time and memory in theory, the actual memory consumption of the suffix tree is very large for large genomes. Suffix arrays (Manber and Myers, 1993) or enhanced suffix arrays (Abouelhoda et al., 2004) have been explored to replace the suffix tree to alleviate this issue, but suffix arrays still require a significant amount of memory usage. To further reduce the memory usage, sparseMEM (Khan et al., 2009) has been developed. In contrast to a full-text suffix array, it stores only a sparse suffix array, i.e. only every K-th suffix is stored. But this can lead to extra computational time in the query phase. Vyverman et al. (2013) further optimizes sparseMEM with a sparse child array that improves running speed without sacrificing the memory usage. Different from these methods that search matches in the forward direction, BackwardMEM (Ohlebusch et al., 2010) constructs a compressed full-text index and uses a backward search method. Similarly, Fernandes and Freitas (2014) employed the backward search technique of the FM-index that works with a sampled representation of the longest common prefix array.
Recently, k-mer index structures are introduced to detect MEMs, as they are easily amenable to parallelization, an important requirement for processing large genomes. E-MEM (Khiste and Ilie, 2015) is currently the most space-efficient method to deal with the problem. It employs a fixed sampling strategy to sample a subset of k-mers at a fixed step on the reference sequence. This sampling method can reduce the number of the indexed k-mers in the hash table for memory saving. However, all k-mers on the query sequence must be generated as keys to search matches in the hash table. The exhaustive query processing is time-consuming. To reduce the number of k-mers from the query sequence, a technique is to use minimizers (Liu et al., 2018; Roberts et al., 2004) to perform a sampling of k-mers on both the reference and query sequences. As suggested by Almutairy and Torng (2018), fixed sampling stores much less k-mers than minimizer sampling does. Although the minimizer sampling method (Roberts et al., 2004) can perform k-mer sampling on two sequences, it only achieves little improvement on querying speed because it produces a number of shared k-mers for an MEM. Very recently, copMEM (Grabowski and Bieniecki, 2019) introduced another sampling technique. The key idea is based on the Chinese remainder theorem to sample k-mers on both sequences R and Q, which is very different from the idea in the minimizer sampling method. Its speed is much faster than the other methods due to the simultaneously reduced number of indexed k-mers and the query k-mers. However, copMEM is unable to detect MEMs of length <50, and there is no multithreading implemented in copMEM.
A common drawback of these k-mer index algorithms is: the index is built only relying on k-mers of the reference sequence, without consideration of the constraint on the target k-mers from the query sequence. For example, a 52-mer (‘GAATTCCTTGAGGCCTAAATGCATCGGGGTGCTCTGGTTTTGTTGTTGTTAT’) occurs twice in the reference genome Homo sapiens. However, it does not occur in the query genome Mus musculus. Insertion of this kind of k-mer into the index consumes extra storage as well as extra time in the search-and-extend step.
To address this issue, Bloom filter is introduced by this work as a bridging point, where k-mers from the reference and k-mers from the query sequence are met in a way that some unnecessary k-mers are filtered to reduce the number of indexed k-mers. In the implementation of this idea, the first step of our algorithm is to perform a fixed sampling to select a subset of k-mers from the query sequence, hence reducing the number of query k-mers. More importantly, these selected query k-mers are stored at a probability data structure Bloom filter. Then all the k-mers of the reference sequence are tested by the Bloom filter. Those k-mers passing the Bloom filter test are inserted into a hash table for indexing. The index construction under the consideration of the target k-mers from the query sequence makes the number of indexed k-mers drastically reduced comparing with the number of sampled k-mers from the reference sequence by E-MEM and copMEM. To our best knowledge, this is the first method that makes connections between the reference k-mers and the k-mers from the query sequences to filter some unnecessary ones before the index construction. Our method is named bfMEM (short form for ‘Bloom filtering for MEM detection’).
We compare the performance of bfMEM with three state-of-the-art algorithms essaMEM (Vyverman et al., 2013), E-MEM (Khiste and Ilie, 2015) and copMEM (Grabowski and Bieniecki, 2019) on five large genomes. Extensive experiments show that our method bfMEM can be two times faster than copMEM with a reduced memory consumption. Our method is also at least 1.8 times faster than E-MEM and one order of magnitude faster than essaMEM.
2 Materials and methods
This section presents definitions and steps for the design of our algorithm bfMEM, then we describe the details of the Bloom filter and rolling hash which are important components in the method.
2.1 bfMEM algorithm
Our algorithm detects all MEMs of length between the reference sequence and the query sequence , where . By convention, we use notation to denote a k-mer (i.e. a substring of length k) starting at the position i of a sequence S. We also add two dummy symbols to R and Q: and . In this way, an MEM is defined as a tuple (i, j, z) satisfying that and , where is the length of the MEM.
It is clear that we can obtain all the MEMs directly by comparing all possible pairs of L-mers from the reference and query sequences. Although a hash table, built on the L-mers of the reference sequence, can avoid some unnecessary comparison, this approach is time-consuming and takes huge memory simply because the number of L-mers from the reference and query genomes is large.
To reduce the number of L-mers in the hash table, intuitively it is good to add all L-mers of Q to a Bloom filter. Then test all L-mers of the reference sequence R at the Bloom filter and only those L-mers passing the test are inserted into a hash table for indexing. Therefore, only a fraction of L-mers of the reference sequence which also occur at the query sequence are stored. In the search-and-extend procedure, every L-mer on the query sequence is used as the key to search in the hash table.
There are still some concerns for this Bloom filtering approach in terms of memory consumption and running speed. (i) The number of L-mers from Q is , linear to the length of Q which is a large number under our assumption. According to Equation (1), the required size of the Bloom filter has to be very large to maintain a low false positive probability (FPP). For example, the query genome M. musculus has number of 80-mers, and the required memory for the Bloom filter is about 3 GB if FPP is set at 0.01. In addition, adding these 80-mers to the Bloom filter is time-consuming. (ii) In the stage of search-and-extend, all the L-mers from the query sequence should be used to search matches in the hash table and later, those matches need to be extended to an MEM. This is time-consuming, let alone the fact that there are many duplicate MEMs. To reduce the memory consumption and to accelerate the matching speed, our idea is to sample less number of k-mers at the very beginning, conditioned that there are no loss of matches at the detection stage.
Our algorithm bfMEM has the following three main steps: (i) sampling k-mers on the query sequence; (ii) construction of an index of k-mers; (iii) MEMs test and generation. A schematic diagram of bfMEM is depicted in Figure 1. The rationale behind bfMEM, that every pair of L-mers is compared, is the same as that of E-MEM. In fact, not to miss any MEMs, the sampling method of the step (i) ensures that all L-mers from the query sequence can be obtained by extending the sampled k-mers; and all k-mers from the reference sequence are compared to the sampled k-mers from the query sequence in the step (ii). These two steps guarantee that all pairs of L-mers on the reference and query sequences are compared via comparing these k-mers. In the second step, the Bloom filter is used to compare a k-mer with a set of k-mers. A property of Bloom filter that false negative matches are impossible guarantees the correctness of filtering k-mers. In our algorithm, the hash values of all k-mers are efficiently computed by the rolling hash function ntHash (Mohamadi et al., 2016). For ease of presentation, we use the notion to represent ntHash hereinafter.
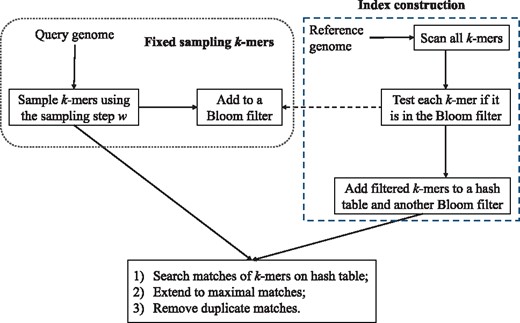
Schematic diagram of our algorithm bfMEM
2.1.1 Fixed sampling of k-mers on the query sequence
To reduce elements to be added to the Bloom filter, we employ the fixed k-mer sampling method from E-MEM (Khiste and Ilie, 2015). Loosely speaking, the fixed k-mer sampling is to select a k-mer common substring of w contiguous L-mers to represent all of them. An example of finding the required common substring is depicted in Figure 2, where w = 8, L = 19. The k-mer common substring is a 12-mer (i.e. k = 12) marked in red which represents the eight contiguous 19-mers.

Fixed sampling of k-mers. In this example, L = 19, k = 12 and w = 8. The red font substring (i.e., the 8th line in the dashed box) is a common substring of the 8 contiguous 19-mers. The underline substrings are 12-mers. The 8-th 12-mer is sampled to represent these 8 19-mers
Assume is the array of all k-mers of the query sequence and . Taking , the sampled k-mers set is , where , that guarantees every L-mers can be obtained by extending the sampled k-mers and minimizes the number of sampled k-mers as well. It is noted that the last k-mer is sampled when w is not a divisor of . It can be seen that the number of sampled k-mers is , less than the number of L-mers of the query sequence. All the sampled k-mers are then added into a Bloom filter . The details are described in Algorithm 1.
If the FPP of is set as 0.01, then the optimal number of hash functions is and the length of bit array of the Bloom filter is . Note that these theoretic settings are the worst-case study, since some sampled k-mers from the query sequence are duplicated, i.e. the number of unique k-mers that are added to fq is smaller than u. For example, the number of sampled k-mers (with ) from the genome of M. musculus is 91 473 837, but the number of k-mers added to the Bloom filter is 86 099 573 (see Supplementary Table S2). With these observations, an actual FPP of is expected to be . The required memory of the Bloom filter is about 108 MB to store all the sampled k-mers when L = 80 and k = 52, while 3 GB memory is required for the Bloom filter to store all the L-mers under the same FPP setting. This is another advantage of the sampling method to handle the query genome M. musculus.
: Sampling k-mers on query sequence

Index construction

2.1.2 Index construction
After the fixed sampling of k-mers from the query sequence, the index is built via the reference sequence and fq. We scan all k-mers (the number of k-mers is about ) in the reference sequence R and test whether they are in . Those k-mers that pass the Bloom filter test are candidate k-mers. Although there is false positive rate, most k-mers in the reference sequence can be filtered by . It is expected that the number of candidate k-mers is much smaller than v. For example, only about one quarter of u k-mers (out of v k-mers) pass the test in the detection of MEMs for the reference genome H. sapiens and the query genome M. musculus (with L = 80 and k = 52). All the candidate k-mers are then inserted into a separate Bloom filter , which employs the same bit array length of . It is worth mentioning that the FPP of is much smaller than 0.01 because, at this stage, the number of filtered k-mers in the reference sequence is much less than u (see FPP analysis in Supplementary Material). Meanwhile, the candidate k-mers are also inserted into a hash table (k-mer as key and its position as value) for indexing as well. The details of the index construction algorithm are summarized in Algorithm 2.
MEMs generation

2.1.3 MEMs generation
As w contiguous L-mers are represented by one k-mer, only the sampled k-mers from Q need to be considered in the final query stage. For a selected k-mer, we first test if it is in . Successfully tested k-mers are then used as the key to retrieve possible left and right extensions by querying . If it has extensions and the length of the extended string is , it is marked as an MEM. The details are listed in Algorithm 3.
For large genomes which are highly similar, it is not surprised that the number of MEMs (L = 40 or 50) is very large. However, copMEM stores MEMs from a subsequence in memory in the implementation. To reduce memory usage, those extensions with length can be written to files like what has done in E-MEM (Khiste and Ilie, 2015). While sorting large temporary file is still time- and memory-consuming. We make another improvement to achieve better time and memory trade-offs. For a big file, we hash matches into some small files and sort them independently to remove duplicates (lines 1 and 2 of Algorithm 3). The lack of full sorting of results leads to different order compared with the results of E-MEM and copMEM. We provide a tool ‘formatconvert’ to reorder the MEMs. After reordering, the order is the same as that of E-MEM and copMEM. The running time and memory usage of the tool are provided in Supplementary Table S4. It is noted that our method uses more disk space than E-MEM and copMEM as we store MEMs in temporary files and divide a large file into small files.
2.1.4 Implementation
To exploit the power of modern multicore computers, the proposed method is implemented with parallel supports, which is also a key advantage of our algorithm over the existing ones as surveyed in Introduction. For the query genome, the algorithm is paralleled over the subsequences. The reference genome is divided equally into pieces according to the number of threads and each thread handles one piece of the reference genome. The hash values of their k-mers are computed and tested by the Bloom filter independently. Additionally, the Bloom filter is implemented as a shared memory structure of multi-threads and adding elements to it is achieved by using atomic operations to control the synchronization. When the Bloom filter is being queried, synchronized accesses are not needed. Finally, removing duplicate matches is paralleled over the small files. It should be noted that bfMEM uses some functions of copMEM, such as read, scan and write functions.
Like some existing tools, our method bfMEM supports forward matching detection (default), reverse-complement matching detection, or both.
2.2 Bloom filter and rolling hash
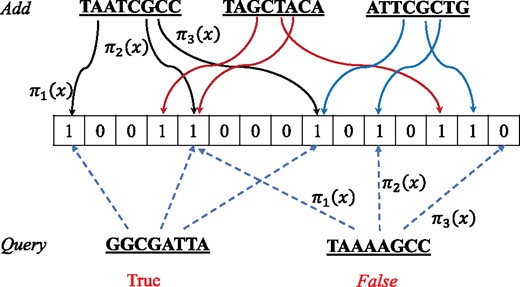
An illustration of adding and querying elements to the Bloom filter
For a given FPP p, the optimal number of hash functions is . In our algorithm and the same as other works (El-Metwally et al., 2016; Song et al., 2014), we adopt p = 0.01 as default.
3 Results and analyses
The performance of bfMEM is assessed by comparing with three state-of-the-art algorithms essaMEM (Vyverman et al., 2013), E-MEM (Khiste and Ilie, 2015) and copMEM (Grabowski and Bieniecki, 2019). All the experiments were carried out on a computing cluster running Red Hat Enterprise Linux 6.7 (64 bit) with 2 × 2.33 GHz Intel® Xeon® E5-2695 v3 (14 Cores) and 128 GB RAM. All tools were run with their default parameters apart from the number of threads. The length parameter L for the minimum length of MEMs ranges from 40 to 300. As essaMEM is very slow under the setting of single thread, we run the program essaMEM with ten threads throughout all the experiments.
3.1 Datasets
Three pairs of reference and query genome comparison experiments, including five large genomes, are performed to evaluate the four algorithms. The datasets are recommended benchmark datasets in the literature (Grabowski and Bieniecki, 2019; Khiste and Ilie, 2015). Supplementary Table S1 provides more information of these five datasets and Supplementary Table S2 lists the setting of reference and query genomes. Hereinafter, we use A versus B to denote that A is the reference genome and B is the query genome in the MEM detection problem.
3.2 Comparison on the number of generated query k-mers
The number of query k-mers also heavily affects the speed of finding MEMs in the search-and-extend stage. As we discussed in Section 1, a drawback of E-MEM is that all k-mers on the query sequence must be used as keys to search matches in the hash table. Differently, bfMEM and copMEM consider only a fraction of k-mers selected from the query sequence, i.e. they make a sampling of k-mers on the query sequence. We compare the numbers of query k-mers used by E-MEM, copMEM and bfMEM in Table 1. E-MEM always processes all the k-mers from the query sequence and the number of query k-mers is at least nine times larger than that of bfMEM. The number of query k-mers used by bfMEM is also at least six times smaller than that of copMEM.
Comparison on the numbers of query k-mers used by E-MEM, copMEM and bfMEM
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | Always | Always | ||||||
| copMEM | N/A | 546 174 368 | 390 124 548 | N/A | 629 074 577 | 449 338 983 | ||
| bfMEM | ||||||||
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | Always | Always | ||||||
| copMEM | N/A | 546 174 368 | 390 124 548 | N/A | 629 074 577 | 449 338 983 | ||
| bfMEM | ||||||||
Note: Bold font indicates the best result in the column. copMEM did not support L < 50.
Comparison on the numbers of query k-mers used by E-MEM, copMEM and bfMEM
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | Always | Always | ||||||
| copMEM | N/A | 546 174 368 | 390 124 548 | N/A | 629 074 577 | 449 338 983 | ||
| bfMEM | ||||||||
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | Always | Always | ||||||
| copMEM | N/A | 546 174 368 | 390 124 548 | N/A | 629 074 577 | 449 338 983 | ||
| bfMEM | ||||||||
Note: Bold font indicates the best result in the column. copMEM did not support L < 50.
3.3 Comparison on the number of indexed k-mers
It is the number reduction of indexed k-mers that can bring obvious benefits for detecting MEMs (Almutairy and Torng, 2018). The reduction brings (i) memory saving for storing the indices; and (ii) query speed up by avoiding more unnecessary matches. In this regard, the second experiment is designed to validate that our algorithm can really reduce the number of indexed k-mers in the final hash table. The numbers of indexed k-mers used by E-MEM, copMEM and bfMEM on the cases H. sapiens versus M. musculus and Triticum aestivum versus Triticum durum are listed in Table 2. The number of indexed k-mers by bfMEM is always one order of magnitude smaller than that of copMEM. On the second case, the performance of bfMEM is better than that of E-MEM for L = 50, 80 and 100. For L = 40 (and k = 32), bfMEM stores more k-mers than E-MEM. This is because (after we investigate further details) there are many duplicate 32-mers on the reference genome T. aestivum (see Supplementary Table S3), and they are added to the hash table independently to avoid missing matches. Nevertheless, bfMEM still runs much faster than E-MEM under the setting of L = 40. The reason is that E-MEM needs to consider all k-mers on the query sequence while bfMEM only processes the sampled k-mers of the query sequence.
Comparison on the numbers of indexed k-mers used by E-MEM, copMEM and bfMEM
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | 261 430 110 | 142 598 242 | 60 330 025 | 43 571 685 | 371 759 667 | 202 778 000 | 85 790 692 | 61 959 944 |
| copMEM | N/A | 743 519 333 | 392 145 164 | N/A | 743 519 333 | 557 639 499 | ||
| bfMEM | 513 266 575 | |||||||
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | 261 430 110 | 142 598 242 | 60 330 025 | 43 571 685 | 371 759 667 | 202 778 000 | 85 790 692 | 61 959 944 |
| copMEM | N/A | 743 519 333 | 392 145 164 | N/A | 743 519 333 | 557 639 499 | ||
| bfMEM | 513 266 575 | |||||||
Note: Bold font indicates the best result in the column. copMEM did not support L < 50.
Comparison on the numbers of indexed k-mers used by E-MEM, copMEM and bfMEM
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | 261 430 110 | 142 598 242 | 60 330 025 | 43 571 685 | 371 759 667 | 202 778 000 | 85 790 692 | 61 959 944 |
| copMEM | N/A | 743 519 333 | 392 145 164 | N/A | 743 519 333 | 557 639 499 | ||
| bfMEM | 513 266 575 | |||||||
| Methods | H. sapiens versus M. musculus | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM | 261 430 110 | 142 598 242 | 60 330 025 | 43 571 685 | 371 759 667 | 202 778 000 | 85 790 692 | 61 959 944 |
| copMEM | N/A | 743 519 333 | 392 145 164 | N/A | 743 519 333 | 557 639 499 | ||
| bfMEM | 513 266 575 | |||||||
Note: Bold font indicates the best result in the column. copMEM did not support L < 50.
3.4 Running time and memory usage comparison
The running time and memory usage for are presented in Tables 3 and 4, respectively. Detailed comparison for L = 150, 200 and 300 are presented in Table 5.
Comparison of running times (in second) by different methods for L = 40, 50, 80 and 100
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 14 735 | 3604 | 1306 | 1130 | 21 660 | 10 284 | 3292 | 2168 | 2493 | 1531 | 797 | 766 |
| copMEM | N/A | 2124 | 221 | 133 | N/A | 8308 | 1168 | 643 | N/A | 1413 | 486 | 338 |
| bfMEM -t 1 | 31 016 | 3678 | 481 | 432 | 46 020 | 14 097 | 2123 | 1020 | 2305 | 813 | 710 | |
| bfMEM -t 5 | 10 916 | 1093 | 146 | 130 | 14 280 | 4725 | 604 | 310 | 618 | 391 | 230 | 190 |
| essaMEM -t 10 | 9362 | 5529 | 3601 | 2656 | 18 048 | 10 662 | 7939 | 5522 | 2121 | 2081 | 1714 | 1819 |
| E-MEM -t 10 | 18 245 | 1824 | 292 | 243 | 25 273 | 7043 | 959 | 616 | 1028 | 628 | 302 | 280 |
| bfMEM -t 10 | ||||||||||||
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 14 735 | 3604 | 1306 | 1130 | 21 660 | 10 284 | 3292 | 2168 | 2493 | 1531 | 797 | 766 |
| copMEM | N/A | 2124 | 221 | 133 | N/A | 8308 | 1168 | 643 | N/A | 1413 | 486 | 338 |
| bfMEM -t 1 | 31 016 | 3678 | 481 | 432 | 46 020 | 14 097 | 2123 | 1020 | 2305 | 813 | 710 | |
| bfMEM -t 5 | 10 916 | 1093 | 146 | 130 | 14 280 | 4725 | 604 | 310 | 618 | 391 | 230 | 190 |
| essaMEM -t 10 | 9362 | 5529 | 3601 | 2656 | 18 048 | 10 662 | 7939 | 5522 | 2121 | 2081 | 1714 | 1819 |
| E-MEM -t 10 | 18 245 | 1824 | 292 | 243 | 25 273 | 7043 | 959 | 616 | 1028 | 628 | 302 | 280 |
| bfMEM -t 10 | ||||||||||||
Note: Parameter ‘-t’ is the number of threads. Bold font indicates the best result in the column. copMEM did not support L < 50 and copMEM did not support multi-threads.
Comparison of running times (in second) by different methods for L = 40, 50, 80 and 100
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 14 735 | 3604 | 1306 | 1130 | 21 660 | 10 284 | 3292 | 2168 | 2493 | 1531 | 797 | 766 |
| copMEM | N/A | 2124 | 221 | 133 | N/A | 8308 | 1168 | 643 | N/A | 1413 | 486 | 338 |
| bfMEM -t 1 | 31 016 | 3678 | 481 | 432 | 46 020 | 14 097 | 2123 | 1020 | 2305 | 813 | 710 | |
| bfMEM -t 5 | 10 916 | 1093 | 146 | 130 | 14 280 | 4725 | 604 | 310 | 618 | 391 | 230 | 190 |
| essaMEM -t 10 | 9362 | 5529 | 3601 | 2656 | 18 048 | 10 662 | 7939 | 5522 | 2121 | 2081 | 1714 | 1819 |
| E-MEM -t 10 | 18 245 | 1824 | 292 | 243 | 25 273 | 7043 | 959 | 616 | 1028 | 628 | 302 | 280 |
| bfMEM -t 10 | ||||||||||||
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 14 735 | 3604 | 1306 | 1130 | 21 660 | 10 284 | 3292 | 2168 | 2493 | 1531 | 797 | 766 |
| copMEM | N/A | 2124 | 221 | 133 | N/A | 8308 | 1168 | 643 | N/A | 1413 | 486 | 338 |
| bfMEM -t 1 | 31 016 | 3678 | 481 | 432 | 46 020 | 14 097 | 2123 | 1020 | 2305 | 813 | 710 | |
| bfMEM -t 5 | 10 916 | 1093 | 146 | 130 | 14 280 | 4725 | 604 | 310 | 618 | 391 | 230 | 190 |
| essaMEM -t 10 | 9362 | 5529 | 3601 | 2656 | 18 048 | 10 662 | 7939 | 5522 | 2121 | 2081 | 1714 | 1819 |
| E-MEM -t 10 | 18 245 | 1824 | 292 | 243 | 25 273 | 7043 | 959 | 616 | 1028 | 628 | 302 | 280 |
| bfMEM -t 10 | ||||||||||||
Note: Parameter ‘-t’ is the number of threads. Bold font indicates the best result in the column. copMEM did not support L < 50 and copMEM did not support multi-threads.
Comparison of memory usage (in GB) by different methods for L = 40, 50, 80 and 100
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| copMEM | N/A | 17.6 | 7.6 | 7.0 | N/A | 43.6 | 10.4 | 8.1 | N/A | 18.5 | 12.7 | 11.2 |
| bfMEM -t 1 | 4.7 | 4.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.6 | 14.6 | 8.5 | 7.8 | ||
| bfMEM -t 5 | 5.1 | 5.0 | 4.9 | 4.9 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.6 | 8.5 | 7.8 |
| essaMEM -t 10 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 9.0 | 9.0 | ||||
| E-MEM -t 10 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| bfMEM -t 10 | 5.1 | 5.7 | 5.6 | 5.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.7 | 8.6 | 7.8 |
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| copMEM | N/A | 17.6 | 7.6 | 7.0 | N/A | 43.6 | 10.4 | 8.1 | N/A | 18.5 | 12.7 | 11.2 |
| bfMEM -t 1 | 4.7 | 4.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.6 | 14.6 | 8.5 | 7.8 | ||
| bfMEM -t 5 | 5.1 | 5.0 | 4.9 | 4.9 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.6 | 8.5 | 7.8 |
| essaMEM -t 10 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 9.0 | 9.0 | ||||
| E-MEM -t 10 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| bfMEM -t 10 | 5.1 | 5.7 | 5.6 | 5.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.7 | 8.6 | 7.8 |
Note: Parameter ‘-t’ is the number of threads. Bold font indicates the best result in the column. copMEM did not support L < 50 and copMEM did not support multi-threads.
Comparison of memory usage (in GB) by different methods for L = 40, 50, 80 and 100
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| copMEM | N/A | 17.6 | 7.6 | 7.0 | N/A | 43.6 | 10.4 | 8.1 | N/A | 18.5 | 12.7 | 11.2 |
| bfMEM -t 1 | 4.7 | 4.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.6 | 14.6 | 8.5 | 7.8 | ||
| bfMEM -t 5 | 5.1 | 5.0 | 4.9 | 4.9 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.6 | 8.5 | 7.8 |
| essaMEM -t 10 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 9.0 | 9.0 | ||||
| E-MEM -t 10 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| bfMEM -t 10 | 5.1 | 5.7 | 5.6 | 5.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.7 | 8.6 | 7.8 |
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | L = 40 | L = 50 | L = 80 | L = 100 | |
| E-MEM -t 1 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| copMEM | N/A | 17.6 | 7.6 | 7.0 | N/A | 43.6 | 10.4 | 8.1 | N/A | 18.5 | 12.7 | 11.2 |
| bfMEM -t 1 | 4.7 | 4.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.6 | 14.6 | 8.5 | 7.8 | ||
| bfMEM -t 5 | 5.1 | 5.0 | 4.9 | 4.9 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.6 | 8.5 | 7.8 |
| essaMEM -t 10 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 6.6 | 9.0 | 9.0 | ||||
| E-MEM -t 10 | 40.2 | 9.1 | 4.8 | 3.8 | 40.2 | 12.2 | 4.9 | 4.0 | 21.1 | 13.2 | 7.0 | 5.7 |
| bfMEM -t 10 | 5.1 | 5.7 | 5.6 | 5.6 | 20.0 | 15.9 | 8.6 | 6.8 | 25.7 | 14.7 | 8.6 | 7.8 |
Note: Parameter ‘-t’ is the number of threads. Bold font indicates the best result in the column. copMEM did not support L < 50 and copMEM did not support multi-threads.
Comparison of running time (in second) and memory usage (in GB) by different methods for L = 150, 200 and 300
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|---|
| L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | |
| E-MEM -t 1 | |||||||||
| copMEM | |||||||||
| bfMEM -t 1 | |||||||||
| bfMEM -t 5 | |||||||||
| essaMEM -t 10 | |||||||||
| E-MEM -t 10 | |||||||||
| bfMEM -t 10 | |||||||||
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|---|
| L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | |
| E-MEM -t 1 | |||||||||
| copMEM | |||||||||
| bfMEM -t 1 | |||||||||
| bfMEM -t 5 | |||||||||
| essaMEM -t 10 | |||||||||
| E-MEM -t 10 | |||||||||
| bfMEM -t 10 | |||||||||
Note: Parameter ‘-t’ is the number of threads. Bold font indicates the best result in the column.
Comparison of running time (in second) and memory usage (in GB) by different methods for L = 150, 200 and 300
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|---|
| L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | |
| E-MEM -t 1 | |||||||||
| copMEM | |||||||||
| bfMEM -t 1 | |||||||||
| bfMEM -t 5 | |||||||||
| essaMEM -t 10 | |||||||||
| E-MEM -t 10 | |||||||||
| bfMEM -t 10 | |||||||||
| Methods | H. sapiens versus M. musculus | H. sapiens versus P. troglodytes | T. aestivum versus T. durum | ||||||
|---|---|---|---|---|---|---|---|---|---|
| L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | L = 150 | L = 200 | L = 300 | |
| E-MEM -t 1 | |||||||||
| copMEM | |||||||||
| bfMEM -t 1 | |||||||||
| bfMEM -t 5 | |||||||||
| essaMEM -t 10 | |||||||||
| E-MEM -t 10 | |||||||||
| bfMEM -t 10 | |||||||||
Note: Parameter ‘-t’ is the number of threads. Bold font indicates the best result in the column.
From the last three rows of Tables 3 and 5 where ten threads are used for all the algorithms, our bfMEM is significantly faster than essaMEM, most of the time even one order of magnitude faster; and bfMEM is at least 1.8 times faster than E-MEM. When only five threads are used by bfMEM, it is still much faster than E-MEM (with ten threads) and is one order of magnitude faster than essaMEM (with ten threads) on most of the cases. It is unfair to compare with copMEM under this scenario as copMEM is only executable on single thread.
When single thread is restricted to run the three algorithms, copMEM (with parameter ) has the best performance in terms of running time. The exception is on the detection of MEMs for T. aestivum versus T. durum with L = 50, where bfMEM is 1.18 times faster than copMEM. In the case of single thread, bfMEM is also much faster than E-MEM except from four cases. That bfMEM is slower than copMEM has the reasons including: (i) the time complexity of computing hash values of all k-mers and querying hash values in the Bloom filter is ; while the time complexity of copMEM’s sampling method is , where k1 is a sampling step; (ii) bfMEM stores and sorts duplicate matches on disk; while copMEM keeps all MEMs in memory. We highlight again that bfMEM is always faster than copMEM and about twice faster on most cases when ten threads are used (copMEM is only executable on single thread).
It is worth noting that short MEMs are very useful for genome comparison due to the diversity of mutations, such as substitution, insertion or deletion of single nucleotides and genome rearrangements, during evolution. Moreover, MEMs with length <100 can be efficiently used for encoding in reference genome compression.
Memory usages of the four algorithms are presented at Tables 4 and 5. All the algorithms use similar amounts of memory when L are not small. In particular, E-MEM is the most frugal when (it is the most space-efficient method as we remarked earlier). However, when L = 40, our method bfMEM and essaMEM save at least six times more than E-MEM on the case H. sapiens versus M. musculus. On the second and third cases (H. sapiens versus P. troglodytes and T. aestivum versus T. durum) with L = 40 and 50, essaMEM uses less memory than other method. When L > 100, essaMEM consumes more memory than E-MEM and bfMEM. For almost all the cases, copMEM uses more memory than the other methods. To conclude, except the fact that copMEM always consumes slightly more memory, all the other algorithms require similar amounts of memory and the actual usage might be data-dependent.
3.5 Effect of multi-threads
Ranging the number of threads from 1 to 10, we show the effect of multi-threads on running time and memory usage of bfMEM in the discovery of MEMs between H. sapiens versus M. musculus with L = 50 or 80. The running speed of bfMEM can be improved 3.5 times if five threads are used, without too much change in memory usage (see details in Supplementary Fig. S1). When more threads are used, the improvement in speed is little. The reason is that the improvement on running time is in trade-off with the I/O time. The memory consumption increases when more threads are used because each thread stores a subsequence in RAM as required by the implementation of bfMEM.
4 Conclusion
In this work, we have proposed a fast algorithm for the MEMs detection problem. Using Bloom filter, bfMEM bridges k-mers from both the reference and query sequence to filter some unnecessary k-mers for reducing the number of indexed k-mers. The experiment results demonstrate that bfMEM significantly outperforms the state-of-the-art algorithms in terms of running time. The superior performance is mainly attributed to the sampling of k-mers on the query sequence and the index construction algorithm. The proposed method cannot be directly used to find maximal approximate matches. In the future, we will investigate whether this index algorithm can be used to discover segmental duplications (Numanagić et al., 2018), periodic repeats (Mori et al., 2019) or other k-mer index-related problems.
Acknowledgements
We thank the anonymous reviewers for their valuable comments and suggestions to improve the quality of our manuscript and source code.
Funding
This work was partly supported by the Australia Research Council Discovery Project ARC DP180100120.
Conflict of Interest: none declared.
References


{kind=link}
{kind=link}
{kind=link}