





Abstract
Interpretation of ubiquitous protein sequence data has become a bottleneck in biomolecular research, due to a lack of structural and other experimental annotation data for these proteins. Prediction of protein interaction sites from sequence may be a viable substitute. We therefore recently developed a sequence-based random forest method for protein–protein interface prediction, which yielded a significantly increased performance than other methods on both homomeric and heteromeric protein–protein interactions. Here, we present a webserver that implements this method efficiently.
With the aim of accelerating our previous approach, we obtained sequence conservation profiles by re-mastering the alignment of homologous sequences found by PSI-BLAST. This yielded a more than 10-fold speedup and at least the same accuracy, as reported previously for our method; these results allowed us to offer the method as a webserver. The web-server interface is targeted to the non-expert user. The input is simply a sequence of the protein of interest, and the output a table with scores indicating the likelihood of having an interaction interface at a certain position. As the method is sequence-based and not sensitive to the type of protein interaction, we expect this webserver to be of interest to many biological researchers in academia and in industry.
Webserver, source code and datasets are available at www.ibi.vu.nl/programs/serendipwww/.
Supplementary data are available at Bioinformatics online.
1 Introduction
An important ingredient to understanding protein function is to identify the interacting residues amongst each other (e.g. Shoemaker and Panchenko, 2007). When compared with the limited number of crystallized structures (Schwede, 2013), a fast growing amount of sequence data is available (e.g. Tuncbag et al., 2008). Given this ever-increasing gap, predicting protein interaction sites from sequence data is an attractive option. To make a widely usable interface predictor, that performs well using only sequence information as input, we recently integrated the following sequence-derived features into a random forest (RF) predictor (Hou et al., 2015).
By implementing these features, we trained our sequence-based protein interface predictors using both homomeric and heteromeric protein interaction datasets. Predictions were significantly more accurate than other predictors on the same test-sets (Hou et al., 2017).
2 The SeRenDIP webserver
The webserver provides a ‘remastered’ version of our previous approach (Hou et al., 2017) which improves the speed of the process by deriving sequence conservation profiles for the homologues by remastering the blast profile of the input sequence (Simossis and Heringa, 2004). The procedure is described in more detail in Supplementary Section S2, see also Supplementary Figure S2. The ‘Remastered’ method is fast enough to allow its practical implementation. The speedup is shown in Supplementary Figure S3.
Based on the features generated with the new approach, we retrain our predictors using the same training and testing protocols as previous research (Hou et al., 2017) to obtain the RF classifiers. More detail is provided in the Supplementary Material. Our new RF models achieve at least the same accuracy, compared with the previous implementation (Supplementary Table S1 and Fig. S5).
For heteromeric interactions, the best option is the RF-hetero predictor. For homomeric, the RF-combined performs better, and it also scores well on heteromeric interactions, making it the best all-round choice. The webserver implements these two final classifiers. As only a single sequence is required as input, and there are no parameters to set, the webserver interface remains nice and clean. A screenshot is presented in Figure 1A. The output is a table of the sequence positions and the corresponding probability score of being an interface site; therefore a score of 0.5 or higher is interpreted as a positive prediction. The higher the probability score, the more confident the prediction is. It is possible to choose different classification thresholds to filter the residues, as can be seen in Figure 1B. We also provide the options to download the raw output file and the full feature table in csv format.
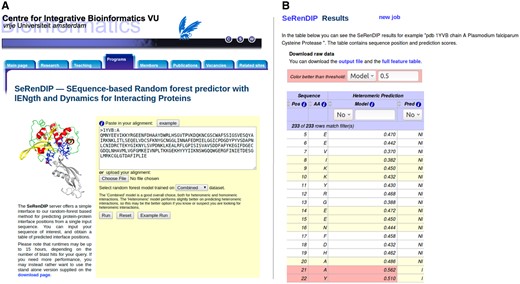
(A) Screenshot of the input form of SeRenDIP. Screenshot of the input page of SeRenDIP available at www.ibi.vu.nl/programs/serendipwww/. The required input is a protein sequence. The only option is to select the Heteromeric or Combined predictor. The heteromeric predictor which scores better on heteromeric proteins than the default Combined. (B) Screenshot of the output page of SeRenDIP. The output is a table with the sequence positions and predictions scores. The first two columns present sequence positions and the corresponding amino acids. The third column includes the corresponding probability score of being part of the interface. The fourth column is the prediction according to the value of the score (‘I’, interface; ‘NI’, non-interface). The predicted interface positions (higher than threshold 0.5) are highlighted in red. Results can be sorted according to different classification thresholds
2.1 Showcase—falciparum cysteine protease
To highlight the impact of accurate interface prediction using our new fast approach, we here show an example of heterodimer protein–protein interaction interface prediction using our webserver.
Falcipain-2 (PDB 1YVB: A) is a cysteine protease from Plasmodium falciparum (Wang et al., 2006). Falcipain-2 interacts with a protease inhibitor, cystatin to form a complex. The protein was not part of our training set, and its sequence identity to any protein in our training data is <25%.
We mapped the predictions from our renewed method and the real interface deduced from the crystal structure of the complex (Supplementary Fig. S1). For this particular interaction, the ‘old’ approach obtained 60.6% coverage and 21.1% precision. The ‘new’ webserver achieved both better coverage of 73.7% of the 33 interface sites, and better precision of 32.4% over the 74 predicted positions. Overall prediction for this target yielded an AUC-ROC of 0.788 and an F1 score of 0.314.
3 Conclusion
The SeRenDIP webserver, for which only a single sequence is needed as input, is correspondingly simple to use. SeRenDIP provides predictions for both homodimeric and heteromeric protein interactions. We therefore expect that the method is immediately applicable in a wide range of biomedical and biomolecular research. The scripts and datafiles are also available as download for stand-alone version.
Conflict of Interest: none declared.
References


{kind=link}