





Abstract
The large-scale kinome-wide virtual profiling for small molecules is a daunting task by experimental and traditional in silico drug design approaches. Recent advances in deep learning algorithms have brought about new opportunities in promoting this process.
KinomeX is an online platform to predict kinome-wide polypharmacology effect of small molecules based solely on their chemical structures. The prediction is made by a multi-task deep neural network model trained with over 140 000 bioactivity data points for 391 kinases. Extensive computational and experimental validations have been performed. Overall, KinomeX enables users to create a comprehensive kinome interaction network for designing novel chemical modulators, and is of practical value on exploring the previously less studied or untargeted kinases.
KinomeX is available at: https://kinome.dddc.ac.cn.
Supplementary data are available at Bioinformatics online.
1 Introduction
By December 2018, 48 small kinase inhibitors have been approved by US FDA, about half of which were approved over the last 5 years. However, the target space or bioactivities of the approved and clinical inhibitors are still surprisingly poorly characterized (Klaeger et al., 2017). Since large-scale biochemical assay to determine kinase targets of compounds are usually costly and time-intensive, the discovery of potent inhibitors with designated selectivity and polypharmacological profile is still a challenging direction in novel drug development process.
Many in silico modelling approaches have been developed to predict kinase inhibitory activity for large-scale compound libraries (Zhong et al., 2018). Compared to traditional drug design methods (e.g. docking and standard QSAR), machine learning-based models such as naïve Bayesian (NB) (Niijima et al., 2012), k-nearest neighbours (KNN) (Stephan and Steven, 2013), random forest (RF) (Bora et al., 2016; Merget et al., 2017), support vector machine (SVM) (Cheng et al., 2012; Niijima et al., 2012) and deep neural network (DNN) (Manallack et al., 2002) have been established to predict a wider range of biological activities for a compound by employing high-dimensional datasets. The separate training process with individual datasets for different tasks of these methods is bound to affect their capability to predict kinome-wide selectivity and polypharmacological profiles. On the other hand, network-aided approaches exploit topological information of drug-target interaction network (Cheng, 2019; Cheng et al., 2019). Assembling targets to be predicted into a network architecture reveals notable advantages of prediction of polypharmacological profiles and drug repositioning, but extending the range of predictive tasks to include tasks with insufficient data could be intractable for network-aided approaches.
Here we present KinomeX, an online platform to predict kinome-wide polypharmacology effect of small molecules based on an established multi-task deep neural network (MTDNN) model. KinomeX implements comprehensive profiling on kinome-wide activity for small molecule, and it can depict the overall selectivity and the selectivity towards a sub-family of kinases based on the predicted kinase profile. We envisage that KinomeX could enable the prediction of kinome-wide polypharmacology effect of small molecules for designing novel chemical modulators or drug repositioning.
2 Main features
2.1 Kinome-wide activity prediction for small molecules based on MTDNN model
The main function of KinomeX is to predict the kinome-wide activity for the small molecules user submitted based on a pre-trained MTDNN classification model. MTDNNs are trained to learn a joint representation of input data points shared for all learning tasks. For the classification model, the representation is fed into N softmax classifiers, one for each task. Previous studies of MTDNN applied to drug discovery suggested that MTDNN can obtain predictive accuracies significantly better than single-task methods for the problems with multiple and related tasks (Ramsundar et al., 2017). Here, over 170 000 bioactivity data points composed of 391 kinases and ∼32 000 compounds were collected from Kinase SARfari database version 6.00 and the Metz dataset (Metz et al., 2011). Eighty percentage of the dataset (∼140 000 data points) were used for training the MTDNN model where one task corresponds to the bioactivity prediction against a specific type of kinase, and rest 20% used for testing (see details in Supplementary Material). ECPF4 was adopted to featurize each molecule and fed into input layer, meaning that only 2D chemical structural information is needed for making prediction based on the resulted model. The outputs of the model are the probabilities whether a small molecule is active against a panel of kinases. A higher probability corresponds to a higher confidence for the predicted interaction. Generally, a predicted interaction with a probability value higher than the cut-off of 0.5 could be considered active. The results can be further narrowed down by setting higher probability cut-offs (0.7 and 0.9) in KinomeX. Extensive computational and experimental validations have been performed. The model shows excellent prediction capability with an auROC of 0.90 on the internal test dataset and consistently outperforms conventional single-task models (Merget et al., 2017) on multiple independent external datasets, especially for the kinases with insufficient activity data (Supplementary Fig. S2 and Table S2).
2.2 Selectivity depiction
Based on the predicted kinome-wide activity, KinomeX also provides a depiction of the overall selectivity and the selectivity towards a sub-family of kinases for the submitted small molecule. Two accepted quantitative evaluation methods, standard score (Karaman et al., 2008) and Gini coefficient (Graczyk, 2007), were adopted to calculate the overall selectivity. To measure of sub-family selectivity, odds ratio (OR) was adopted (Bland and Altman, 2000), which can be considered as the strength of the association between an inhibitor and a sub-family. If the OR is significantly >1.0, it indicates that the inhibitor is selective to the sub-family.
3 Case study
A rigorous experimental validation was performed to investigate the practical benefits of the model for drug discovery or repurposing applications. NVP-BHG712 has been reported as an EphB4 receptor inhibitor showing activity against c-Raf, c-Src and c-Abl (Martiny-Baron et al., 2010). NPV-BHG712 is not included in our modelling dataset, and complete kinase spectrum of it is unavailable. The prediction for activity and selectivity of NVP-BHG712 was performed by KinomeX, and it shows that its reported primary targets can be accurately identified. Besides, NVP-BHG712 was predicted to inhibit other important therapeutic targets, including Src family kinases (Lyn, Hck, Lck and Yes), DDR1 and TrkC, which may implicate the anti-cancer potential of NVP-BHG712. The commercial KinaseProfiler Service (Eurofins Scientific, Inc.) was utilized to evaluate the experimental activity against a panel of 405 kinases. Overall, KinomeX produced high-quality predictions generally in agreement with experiment data with an auROC of 0.86, and the novel ‘off-target’ activities have been confirmed by subsequent experiments at lower compound concentrations (Supplementary Fig. S3 and Table S3).
4 Implementation
KinomeX is freely accessible for non-commercial users in a webserver (https://kinome.dddc.ac.cn). A typical kinome-wide virtual profiling workflow of KinomeX is as following (Fig. 1): (i) A molecule is submitted from user-side (browser) by drawing online (Bienfait and Ertl, 2013) or by uploading (Molecule Submission); (ii) The kinome-wide activities are computed by server-side based on a pre-trained MTDNN model (Activity Computation); (iii) Figures are generated based on the profiling, including a chemical depiction, a kinase phylogenetic tree (Chartier et al., 2013) and selectivity bar charts (Figure Generation); (iv) esults download and/or display online (Display & Download).
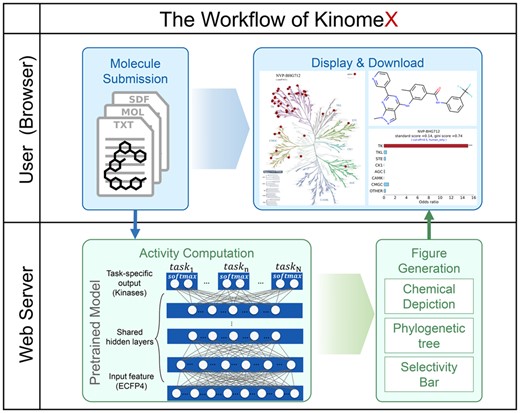
The workflow of KinomeX
MTDNN model was developed with Tensorflow (Version 1.6.0) and DeepChem (Version 2.1.0). For the backend of KinomeX, it uses a MySQL database for data storage and an nginx software as the server engine. For the frontend, it deploys several JS/React modules which support the functionalities such as molecule submission, online display, etc. The main frame of KinomeX is developed within a Django framework, with some backend services written in Python scripts for tasks such as job management, log management, etc. The key features of KinomeX have been fully modularized for better usage and construction. More secure connections to KinomeX have been provided through HTTPS protocols for many popular web browsers. In addition to these, KinomeX is deployed on an AliYun cloud server, which enables better stability and scalability for managing computing resources.
Funding
We gratefully acknowledge financial support from the National Natural Science Foundation of China [81773634 to M.Z.], National Science & Technology Major Project ‘Key New Drug Creation and Manufacturing Programme’, China [2018ZX09711002 to H.J.] and the ‘Personalized Medicines—Molecular Signature-based Drug Discovery and Development’, Strategic Priority Research Programme of the Chinese Academy of Sciences [XDA12050201 to M.Z.].
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, Zhaojun Li and Xutong Li authors should be regarded as Joint First Authors.


{kind=link}