





Abstract
MicroRNAs (miRNAs) are small RNA molecules (∼22 nucleotide long) involved in post-transcriptional gene regulation. Advances in high-throughput sequencing technologies led to the discovery of isomiRs, which are miRNA sequence variants. While many miRNA-seq analysis tools exist, the diversity of output formats hinders accurate comparisons between tools and precludes data sharing and the development of common downstream analysis methods.
To overcome this situation, we present here a community-based project, miRNA Transcriptomic Open Project (miRTOP) working towards the optimization of miRNA analyses. The aim of miRTOP is to promote the development of downstream isomiR analysis tools that are compatible with existing detection and quantification tools. Based on the existing GFF3 format, we first created a new standard format, mirGFF3, for the output of miRNA/isomiR detection and quantification results from small RNA-seq data. Additionally, we developed a command line Python tool, mirtop, to create and manage the mirGFF3 format. Currently, mirtop can convert into mirGFF3 the outputs of commonly used pipelines, such as seqbuster, isomiR-SEA, sRNAbench, Prost! as well as BAM files. Some tools have also incorporated the mirGFF3 format directly into their code, such as, miRge2.0, IsoMIRmap and OptimiR. Its open architecture enables any tool or pipeline to output or convert results into mirGFF3. Collectively, this isomiR categorization system, along with the accompanying mirGFF3 and mirtop API, provide a comprehensive solution for the standardization of miRNA and isomiR annotation, enabling data sharing, reporting, comparative analyses and benchmarking, while promoting the development of common miRNA methods focusing on downstream steps of miRNA detection, annotation and quantification.
https://github.com/miRTop/mirGFF3/ and https://github.com/miRTop/mirtop.
Supplementary data are available at Bioinformatics online.
1 Introduction
microRNAs (miRNAs) are the best known class of small RNAs and were discovered in the nematode worm Caenorhabditis elegans (Bartel, 2004, 2018). It was first reported that the gene lin-4 generated a 22 nucleotide (nt) long RNA molecule that bound to the 3’-UTR of the lin-14 gene transcript, thereby regulating its expression during larval development (Lee et al., 1993). miRNA genes are transcribed into a primary RNA (pri-miRNA) that is processed into a hairpin-like miRNA precursor (pre-miRNA) after cutting off the 5’ and 3’-tails by Drosha and DGCR8 proteins (Denli et al., 2004). The pre-miRNA hairpin is then exported to the cytoplasm and processed by Dicer, which cleaves off the hairpin loop and releases a miRNA duplex about 22 nt long (Perron and Provost, 2008). Originally, it was believed that only one strand of the duplex is retained and incorporated into the RNA-induced silencing complex thereby mediating gene silencing by imperfect base pairing between the miRNA and the 3’-UTR of target messenger RNAs (mRNA) (Vella et al., 2004). It was later shown that both arms of a miRNA can produce mature miRNAs, either simultaneously (Yang et al., 2011), or in a tissue specific manner (Londin et al., 2015; Telonis et al., 2015). In addition, it was shown experimentally that both amino acid-coding sequences (Tay et al., 2008) and 5’-UTRs (Zhou and Rigoutsos, 2014) can also be targeted by miRNAs. miRNAs are essential to virtually all biological processes including, but not limited to, cell differentiation, cell proliferation, cell death, fat metabolism and neuronal cell fate (Bartel, 2004, 2018). Moreover, the deficit or excess of miRNAs have been associated with several human diseases, such as myocardial infarction and different types of cancer (Ardekani and Naeini, 2010). miRNAs reside not only inside cells, but also in a variety of biofluids (Zhang et al., 2018; Zhou et al., 2018), which led to the suggestion that they could be used as non-invasive disease biomarkers or even therapies (Liu et al., 2014; Pan et al., 2018; Telonis et al., 2015; Zhang et al., 2018, 2019).
IsomiRs are sequence variants from annotated miRNAs (Desvignes et al., 2015; Fromm et al., 2015; Kim et al., 2019). IsomiRs were first described by Morin et al. (Morin et al., 2008) in human stem cell lines using next generation sequencing technologies. Sequence variations can affect different parts of the mature miRNA sequence as consequences of different biochemical processes (Pantano et al., 2010). Variations at the 5’ and 3’-ends can be due to imprecision of the Drosha/Dicer cutting machinery(Bofill-De Ros et al., 2019b; Gu et al., 2012). Moreover, it has already been shown that, in humans, these endpoint variations may differ in both healthy individuals and patients (Loher et al., 2014; Telonis et al., 2015; Telonis and Rigoutsos, 2018). In fact, isomiRs are likely genetically controlled because they depend on a person’s sex, population origin and ethnicity (Loher et al., 2014; Telonis et al., 2015), as well as on tissue, tissue state and disease subtype (Magee et al., 2018; Telonis et al., 2017). Non-templated nucleotide additions at the 3’-end can be due to terminal uridylyl transferases that generally add adenine or uridine nucleotides (Menezes et al., 2018). Variations and non-templated additions at the 3’-end can assume a new function considering that the tail end of a miRNA can contain a cell-compartment localization signal (Hwang et al., 2007) or change target-specificity (Yang et al., 2019). Finally, post-transcriptional processing of miRNAs can generate nucleotide changes at any position of the sequence by RNA processing enzymes, such as A-to-I editing by RNA-specific adenosine deaminase (ADAR) enzymes (Kawahara et al., 2007). The specific function of isomiRs is still not well understood, but multiple studies have suggested a context-specific effect of isomiRs on gene regulation (Garate et al., 2018; Menezes et al., 2018; Telonis et al., 2017; Trontti et al., 2018). This conclusion is further supported by the fact that different isomiRs from the same mature miRNA can target virtually non-overlapping sets of transcripts (Engkvist et al., 2017; Kume et al., 2014; Tan et al., 2014; Telonis et al., 2015; Yang et al., 2019).
Several tools have been developed to analyze miRNAs and their respective isomiRs (Lukasik et al., 2016) (Supplementary Table S1). These tools differ in their alignment strategies, ways to handle cross-mapping events, abundance cutoffs, or isomiR annotation methods. Many tools operate by mapping sequenced reads on a curated database such as MirGeneDB (https://doi.org/10.1101/258749), miRBase, miRCarta, or RNACentral (Backes et al., 2018; Fromm et al., 2015; Kozomara and Griffiths-Jones, 2014; Sweeney et al., 2019). Some tools allow users to provide custom database of interest: e.g. species-specific annotation, all members of the let-7 family, miRNA precursors or shRNA products. The benefit of such an approach is its speedy execution due to the small size of the search space. Databases like isomiR Bank, re-analyze public datasets and share the annotation through a web-page (Zhang et al., 2016). Each of these tools report isomiRs in a different format and with different levels of complexity (Supplementary File S1).
Seqbuster integrates its own aligner to maximize the number of isomiRs analyzed but only retains isomiRs with a maximum of one nucleotide change within the miRNA (missing the cases with more changes) and three different nucleotides at each end (Pantano et al., 2010). Seqbuster outputs a tabular delimited file with a column for each isomiR type. It works with the isomiR Bioconductor package to detect expression data and isomiR differences (https://doi.org/doi: 10.18129/B9.bioc.isomiRs).
isomiR-SEA (Urgese et al., 2016) implements a miRNA-specific alignment procedure for comparing each read of the sample to all the miRNA sequences from miRBase and MirGeneDB, collecting uniquely and multi-mapped sequences (Fromm et al., 2015; Kozomara and Griffiths-Jones, 2014). This tool annotates the positions of the variations (mismatches and indels) enabling fine categorization of each aligned read that can be classified as canonical miRNA or one of the isomiRs described in Supplementary Tables S2 and S3. isomiR-SEA then outputs a detailed isomiR expression quantification table, with a focus on the conserved miRNA-mRNA interaction sites. isomiR-SEA is implemented in C++ using functions collected in the SeqAn bioinformatics library (Reinert et al., 2017).
sRNAbench (Aparicio-Puerta et al., 2019) applies a bowtie seed alignment option (Langmead et al., 2009), either to the genome (genome mode) or to miRNA reference sequences (library mode), to score only the first L nucleotides (by default L = 19 allowing one mismatch) and therefore does not take into account mismatches at the 3’-end of the read caused by any post-transcriptionally added nucleotides. sRNAbench clusters all reads that map to the reference precursor within a window of the canonical mature miRNA sequence (3 nt upstream of the start coordinate and 5 nt downstream of the end coordinate) and applies a hierarchical isomiR classification scheme. The sRNAbench tool has several tab-separated output files for isomiR analysis.
miRge2.0 maximizes isomiR discovery by iteratively mapping reads to user-defined miRNA and non-miRNA libraries using bowtie with a final step of loose alignment to the miRNA reads of any unaligned sequences (Lu et al., 2018). miRge2.0, has a threshold option to remove called miRNAs whose reads are predominantly isomiR, rather than canonical, based on a user-specified threshold to correct for false positive miRNAs.
Prost! (PRocessing Of Small Transcripts) quantifies and annotates miRNA expression (Desvignes et al., 2018). Prost! uses the global aligner BBMap (https://sourceforge.net/projects/bbmap/) to align transcripts to a user-specifiable genome assembly allowing for the identification of post-transcriptional modifications (e.g. non-templated additions, editing, alternative cutting) as well as identifying whether an isomiR can equally likely originate from one or more genomic loci. Prost! then groups transcripts based on genomic location(s) and each group of sequences is annotated with user-defined databases of mature miRNAs, miRNA precursors and other types of RNAs. Genomic location groups with identical annotations are further combined and can be used for downstream differential expression analyses.
IsoMiRmap maps and quantifies isomiRs by considering both a miRNA library and the genome (ignoring miRNAs region). The IsoMiRmap tool (Loher and Rigoutsos––Personal Communication), currently in development, considers the entire genome when mapping while having modest computational requirements. Considering the entire genome has the advantage of being able to flag whether or not an isomiR is exclusive to the miRNA library or if it could have been transcribed from a gene different from that of the canonical miRNA sequence. The IsoMiRmap tool outputs in various formats, including HTML, tab separated files and mirGFF3.
OptimiR produces miRNA and isomiR expression abundances, and optionally integrates genetic information to retain or discard alignments depending on their consistency with the genotype of the sample (Thibord et al., 2019). If the user provides a vcf file with genetic variants located on mature miRNAs, the miRBase reference library is automatically updated with new sequences that integrate the variants. The alignment procedure relies on bowtie2 local alignment mode, without any mismatch allowed in the central sequence (Langmead and Salzberg, 2012). A customizable score is then computed for each alignment, which resolves cross-mapping events and discards unreliable alignments.
Although the analysis of miRNAs and their isomiRs has dramatically changed over the past several years, a lack of consensus persists among bioinformatic tools used to describe and study the isomiR landscape. Tools generate different output file types with different structures and isomiR notations. This lack of homogeneity, which has an advantage in representing a diversity of ways of approaching isomiRs, however, prevents the evaluation of each tool to case-specific situations and precludes data sharing and the development of common downstream analyses that would be independent of the tool used for detection and quantification.
To overcome this situation, we present here mirGFF3, a standardized output format for the analysis of miRNAs and their isomiRs based on transcriptomic sequencing data. mirGFF3 was created to fit all research fields and as many tools as possible with the idea of democratization and standardization of data analysis. This new file format allows the storage of relevant miRNA/isomiR information and was developed based on the existing GFF3 (General Feature Format) format (https://github.com/The-Sequence-Ontology/Specifications/blob/master/gff3.md), commonly used in genome annotation and mRNA analyses. Importantly, mirGFF3 uses an ontological naming system to relate identified sequence features to the sequence ontology project (Eilbeck et al., 2005). Moreover, we developed a Python API (mirtop) that supports general file operations as well as importing miRNA tool output files and converting and exporting them into the new mirGFF3 format to promote the development of downstream tools usable by all in a collaborative environment.
2 Results
To communicate ideas, define standards, and to develop successful formats and tools useful to the majority of researchers in the miRNA community, we created the miRNA Transcriptomic Open Project (miRTOP), an entirely open source and community-based project. The project is open for participation to any member of the miRNA community, regardless of level of seniority and status. miRTOP serves as an incubator of ideas that helps improve miRNA analysis standards and boost collaboration. All updates and progress reports are and will continue to be publicly available as they happen, and discussion summaries have already been released through GitHub. The project owns its own GitHub organization which articulates so far four different repositories: (1) the main web page, (2) the mirGFF3 format, (3) the mirtop API and (4) the incubator (https://github.com/miRTop/incubator/issues), where new ideas take form. Additional repositories will be added as projects develop. The miRTOP group uses the GitHub project web pages to organize different analyses in a transparent, communicative and inclusive manner to promote collaboration and equality among all members of the miRNA community. Crowd-supported projects have recently started to emerge in bioinformatics research (Lesurf et al., 2015). As one of them, miRTOP encourages communication, collaboration and community-driver problem solving as well as decision making. The research problems are selected by the miRNA community and commonly addressed.
2.1 The mirGFF3 file format: definition and explanation
The mirGFF3 format was developed based on the original GFF3 format, taking advantage of the coordinate system information that GFF3 can handle and the possibility to store attributes in column 9 (Supplementary File S2). The GFF3 format is commonly used for the annotation of genomic coordinates and is a popular data exchange format, particularly within the Generic Model Organism Database (O’Connor et al., 2008) and genome browsing applications such as Ensembl or IGV (Thorvaldsdottir et al., 2013; Zerbino et al., 2018). The mirGFF3 format definition and corresponding descriptions are maintained on the mirGFF3 specific GitHub page, and have been deposited in the FAIRsharing (Sansone et al., 2019) and EDAM databases (Ison et al., 2013). Other file formats, such as BED, BAM, or VCF files, were considered but their customization to miRNA data would have necessitated more complicated alterations to be unbiased compared to the adaptation of the original GFF3 file format. For instance, many extra columns would have been necessary to adapt the BED file format to define miRNA attribute information, and for BAM and VCF files, several different isomiR attribute tags would have to be implemented in addition to the already mandatory ones. In contrast, the original GFF3 file format already provides a structure fulfilling all the miRNA and isomiR requirements without the need to create a totally new file format or extension.
In the mirGFF3 format, the columns ‘seqid’, ‘source’, ‘type’, ‘start’, ‘end’ and ‘strand’, are used as defined in the original GFF3 format (Supplementary Table S2). The column ‘type’ accepts the terms ‘ref_miRNA’ or ‘isomiR’ which are part of the sequence ontology project for miRNA definition (http://www.sequenceontology.org/browser/current_release/term/SO: 0000276) as SO: 0002166 and SO: 0002167, respectively. The column ‘score’ is available for each tool to use freely if additional information needs to be added or specified. The column ‘phase’ is ignored in the mirGFF3 format given that it refers specifically to reading frame in protein coding sequences. Finally, column 9, ‘attribute’, was adapted to contain all the relevant information concerning the metadata that characterize each specific isomiR (Supplementary Table S3). In the mirGFF3 definition, attributes starting with a capitalized letter are reserved to the attributes listed in Supplementary Table S3, but custom attributes can be added by adding their descriptor in lower case.
mirGFF3 format accepts headers that include sample origin, names and other custom information used to parse the data by the API framework. All header lines should start with the string ‘##’. Four header lines are mandatory: (1) the mirGFF3 format version, (2) the database used for annotation, (3) the sample names and (4) the tool used for annotation and quantification (Fig. 1a). The database line can point to any of the already published resources and their version: miRBase (Kozomara and Griffiths-Jones, 2014), miRCarta (Backes et al., 2018b), miRGeneDB (Fromm et al., 2015), or a custom database. For existing databases, the version should be provided. For custom databases, an optional link to download the coordinates or precursor sequences should be provided. Sample names should be given after the character string ‘COLDATA’ and should contain the sample names, each separated by a ‘,’ (comma) character (if more than one sample). The header string, ‘TOOLS’, is used to inform the tool used to detect miRNAs and isomiRs from the transcriptomic sequencing data. If the attribute ‘Filter’ is used, a line starting with the character string ‘FILTER’, which explains the possible values this attribute refers to, should be added for the user to filter the file content based on these criteria (Fig. 1a). In addition, we encourage users to add any header line that could provide additional useful information concerning the original small RNA-seq analysis. The most common information could be, but is not limited to, the command line/parameters used to generate the mirGFF3 file, the date of generation, or the description of any custom adjustments done during the previous annotation and quantification analysis.
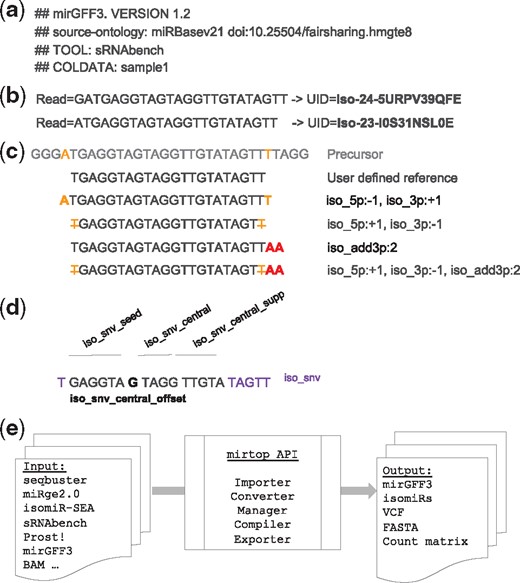
The mirGFF3 file format and the mirtop API. (a) Example of input file header and the required lines: file format version, database used and samples included. (b) Examples of sequence compression to uniquely identify each sequence. (c) Examples of isomiRs with changes at the 5’- and 3’-ends and their respective variant attributes. The first sequence represents a portion of the miRNA precursor (i.e. pre-miRNA) and the second sequence is the reference isomiR defined by the user or the database used. Bold orange letters indicate templated additions. Bold red letters indicate non-templated additions at the 3’-end of the isomiR. Orange strikethrough letters indicate non-templated nucleotide additions at the 3’-end of the isomiR. (d) Example of nucleotide changes at different positions/regions of the isomiR and their respective naming. (e) The mirtop API workflow shows the main formats accepted as input files, the functions the Python API have implemented and the output file formats
The column ‘attribute’ (column 9) of the original GFF3 format was adapted to contain all the metadata relevant to miRNA/isomiR analyses [e.g. the exact sequence variation(s) that an isomiR displays, its expression in different samples, its mapping location] with the possibility to filter and classify isomiRs by mature miRNA(s) and/or pre-miRNAs(s) (Supplementary Table S3). The unambiguous identification of isomiRs between studies is still an open problem because different tools utilize different nomenclature and categorization systems. To this end, we adopted a unique identifier (UID), or ‘IsomiR license plate’, inspired by the MINTmap approach for tRNA fragments (Loher et al., 2017; Pliatsika et al., 2016) and acting as a sequence-dependent unique ID that is independent of genome assembly or species and does not require an arbitrary naming mechanism. Any isomiR sequence can be translated into a UID and any UID can be converted back to the isomiR sequence it represents (Fig. 1b). In the mirGFF3 format, a UID name, e.g. ‘iso-NN-C{N}’, concatenates three pieces of information, each separated by a ‘-’ (dash). First, the prefix ‘iso-’ specifies that this sequence corresponds to an isomiR; second, the length (NN) of the sequence is provided and third, the suffix is the encoded nucleotide sequence. The nucleotide sequence conversion method follows the rules of MINTmap in which each nucleotide triplet is encoded into a single character therefore reducing the string length by one third.
The ‘Variant’ attribute constitutes another characteristic of the mirGFF3 format specifically organized to maximize clarity, communication and standardization across the community. The ‘Variant’ attribute follows an isomiR description and miRNA–mRNA interaction characteristic adapted from the isomiR-SEA format (Urgese et al., 2016). Briefly, isomiR modification characterizations are based on a comparison of the sequence of a given isomiR to its reference miRNA in the chosen database. Changes on the 5’-end of the sequence, related to the start of the miRNA, are described as ‘iso_5p’ and changes on the 3’-end of the sequence, related to the tail of the miRNA, are described as ‘iso_3p’. This annotation prefix is followed by details on the nucleotide changes of this isomiR compared to the provided reference. A ‘–’ (minus sign) is used if the isomiR start or end is upstream compared to the reference extremity. In contrast, a ‘+’ (plus sign) is used if the isomiR start or end is downstream compared to the reference endpoint (Fig. 1c) (Loher et al., 2014; Telonis et al., 2015). For example, an isomiR with both ‘iso_5p:–1’ and ‘iso_3p:+1’ as ‘Variant’ attributes would be two nucleotides longer than its reference: one nucleotide longer at the 5’-end and one nucleotide longer at the 3’-end (Fig. 1c). In both ‘iso_5p’ and ‘iso_3p’ cases, the nucleotide additions have to be templated additions, meaning that these nucleotides are encoded in the genome. In cases of 5’-non-templated additions (additions that do not match the reference genomic sequence), isomiRs are described as ‘iso_add5p’ (Fig. 1c). Similarly, in the case of 3’-non-templated additions, isomiRs are described as ‘iso_add3p’.
Finally, isomiRs that present nucleotide changes in their sequence that do not affect their extremities are described as ‘iso_snv’ (single nucleotide variant). This type of isomiR is further divided into five subtypes (Fig. 1d): (1) ‘iso_snv_seed’, when the nucleotide variation is located in the seed of the detected isomiR between nucleotides 2 to 7; (2) ‘iso_snv_central_offset’, when the nucleotide variation is located at the seed offset position, at nucleotide 8, a nucleotide that is relevant to the strength of the miRNA-mRNA interaction; (3) ‘iso_snv_central’, when the nucleotide variation is located in the central part of the miRNA, between nucleotide 9 to 12, (4) ‘iso_snv_central_supp’, when the nucleotide variation is located in the supplementary region of the miRNA, between nucleotides 13 to 17 and (5) ‘iso_snv’, when the nucleotide variation is located in any other position in the miRNA, nucleotides 1, and 18 to the end of the miRNA.
The ‘Filter’ attribute was adapted from the variant caller format file, where it is used to decide whether a variant passes or not the user-defined filtering options. Each annotation and quantification tool has the possibility to attribute to each isomiR a reliability score that can be any custom value defined in the additional header lines of the mirGFF3 file.
The ‘Hits’ attribute is used to represent the number of times that the read name/sequence matches the database with different isomiR changes. For example, in the human genome assembly GRCh38 (International Human Genome Sequencing Consortium, 2004), iso-22-DV0Y6O6N3 (with sequence AATGCACCTGGGCAAGGATTCT) can be attributed to both MIMAT0002871&hsa-miR-500a-3p (ATGCACCTGGGCAAGGATTCTG) and MIMAT0004775&hsa-miR-502-3p (AATGCACCTGGGCAAGGATTCA) with different but presumably equally likely variations from the two references. In the first assignment case (hsa-miR-500a-3p), the 5’-end and 3’-end differ from the reference by one nucleotide, whereas in the second assignment case (hsa-miR-502-3p), the 3’-end differs from the reference by the insertion of one nucleotide. By setting ‘Hits = 2’ and representing the sequence in two lines (with ‘Parent’ attribute being one of the references in each line), both possible origins can be adequately captured. The ‘Expression’ value for the variant is set to the number of total reads for the sequence, and not a proportion of them, and the ‘UID’ attribute can be used to parse the file and avoid over-counting. A different example could be the isomiR iso-23-UPVMX5I80O (with sequence TACAGTAGTCTGCACATTGGTTA) that can be attributed to three different loci located on three different human chromosomes: MIMAT0004563&hsa-miR-199b-3p on chromosome 9 and MIMAT0000232&hsa-miR-199a-3p on chromosomes 1 and 19 (all three loci having ACAGTAGTCTGCACATTGGTTA as reference sequence). In this situation, one can set ‘Hits = 3’ and take a similar approach as above. Alternatively, because this isomiR perfectly matches each genomic location in the exact same way, it could be listed in a single line with the ‘Hits’ attribute set to ‘1’ and the ‘Parent’ attribute would be used to reflect the multiple possible origin by having the three reference names separated by a comma character.
We realized that column 9 can be overwhelmed by the number of attributes it contains; for that reason, a mirGFF3 file can be converted into a tabular format facilitating the parsing by other tools or custom scripts. In addition, mirGFF3 can be output as a GTF format changing the separator character used in the ‘Attributes’ column.
2.2 The mirtop API framework
The API framework ‘mirtop’ was developed in Python (v.2.7 and v3.6) and uses other common bioinformatics packages. It operates BAM files (pysam) (Li et al., 2009), Bed files (pybedools, bedtools) (Dale et al., 2011; Quinlan, 2014) and standard IO processes with sequences (Biopython) (Cock et al., 2009). The mirtop package is based on a central class that converts each line of the mirGFF3 file into a Python class structure, containing all the information related to each isomiR. A validation step for mirGFF3 rules and restrictions occurs at the creation of the file, avoiding errors that can be difficult to uncover later.
The mirtop API framework contains five different operations: importing, converting, managing, compiling and exporting (Fig. 1e). The importers in mirtop have so far been coded to import and convert the output files of seqbuster (bcbio-nextgen), miRge2.0, isomir-SEA, sRNAbench and Prost! into the mirGFF3 format. Furthermore, IsoMiRmap, miRge2.0, OPTIMIR and QuagmiR (Bofill-De Ros et al., 2019a) have already implemented the mirGFF3 format into their outputs. This is an indication of the short adaptation time required thanks to the use of the standard GFF3 format mirtop uses. The mirtop operator can also manage and compile mirGFF3 files allowing joining, filtering on single or multiple files and transformation of the mirGFF3 information into a count matrix. Finally, mirtop exporters create the final mirGFF3 file and can also convert it into other output formats commonly used for downstream analyses. Currently, mirtop, in addition to the mirGFF3 format, can export to FASTA, isomiRs (Bioconductor package, https://bioconductor.org/packages/release/bioc/html/isomiRs.html) and VCF formats, which are all used in a diversity of visualization and analysis tools for isomiR characterization and variant calling (http://www.internationalgenome.org/wiki/Analysis/vcf4.0/).
The conversion of several different tool outputs into a common file format will help researchers and developers focus on downstream analyses without being limited to only one quantifying tool and a specific output format. The mirtop API will therefore help boost the development of universal downstream analyses, enhancing the reproducibility and quality of miRNA and isomiR biology research.
3 Discussion
Here, we present a community-backed effort to standardize, homogenize and enhance the ways researchers report, share and communicate miRNA results. We have organized a community with a common goal for miRNA/isomiR result standardization, and created mirGFF3, an adapted GFF3 file format. mirGFF3 was specifically designed to contain all relevant information concerning miRNAs and isomiRs identified in small RNA-seq data, regardless of the upstream methods or downstream use-cases. This new format represents the first consensus supported by multiple experts in the field for the report of isomiR variations and abundances in one or more biological samples produced by high throughput sequencing technologies. The mirGFF3 format is complementary to existing bioinformatics tools that support GFF3 files and aligns to the transcriptomic communities that have based their mRNA annotations on GFF3 files. Similar to BAM or VCF file formats, mirGFF3 contains all the information necessary to re-analyze the data in the same way as when the raw output file from any analysis pipeline is available. The API framework, mirtop, which enables the conversion of miRNA quantification tool outputs and the processing of general statistics and count matrices, will serve as a catalyst for the use of the mirGFF3 format. The mirtop API supports any version of the mirGFF3 format and can convert older files to the latest version if needed.
The mirGFF3 file format and the mirtop API tool are the results of an open-membership international miRNA community created to promote open source code sharing in a collaborative and well-supported bioinformatic environment. The mirGFF3 format and associated mirtop API will encourage the miRNA community to develop downstream analysis protocols independent of the initial tool that was used for detection and quantification. The mirGFF3 format will provide a common entry point for a variety of applications ranging from the annotation of miRNAs/isomiRs or filtering for technical errors inherent to each library preparation protocol (Giraldez et al., 2018), to visualization, variant calling, differential expression, clustering, or any other sequence analyses.
We are currently using mirGFF3 and mirtop to study the accuracy of isomiRs detection across laboratories, protocols and tools by re-analyzing multiple publicly available datasets (Giraldez et al., 2018; Kim et al., 2019; Wright et al., 2019). The current status of this project can be accessed at: https://github.com/miRTop/isomir_accuracy_meta_analysis. The use of the mirGFF3 format and mirtop makes comparisons easier, more transparent and reproducible.
The miRTOP group is and will remain open to any researcher interested in small RNA analysis at any level, from experimental scientists to computational biologists. miRTOP was created by members of the miRNA research community for the miRNA research community and offers networking and organization to improve and to promote collaborative research.
Acknowledgements
Authors thank Peter Batzel for suggesting to us adapting the GFF3 format, Rafael Alis for helping in the debugging the mirGFF3 conversion function, Yin Lu for integrating mirGFF3 into miRge2.0 and Shruthi Bandyadka for integrating the tabular exporter operation.
Funding
This work was supported by grant PLR-1543383 and OPP-1543383 of the National Science Foundation (T.D. and J.H.P.), B.F. and M.R.F. acknowledge funding from the Strategic Research Area (SFO) program of the Swedish Research Council (VR) through Stockholm University, M.K.H. was supported by grant 1R01HL137811 of the National Institutes of Health, National Heart Lung Blood Institute, F.T. was financially supported by the GENMED laboratory of excellence on medical genomics (ANR-10-LABX-0013) and I.S.V. was supported by the George and Marie Vergottis Fellowship of Harvard Medical School.
Conflict of Interest: none declared.
References


{kind=link}