





Abstract
RNA structure is difficult to predict in vivo due to interactions with enzymes and other molecules. Here we introduce CROSSalive, an algorithm to predict the single- and double-stranded regions of RNAs in vivo using predictions of protein interactions.
Trained on icSHAPE data in presence (m6a+) and absence of N6 methyladenosine modification (m6a-), CROSSalive achieves cross-validation accuracies between 0.70 and 0.88 in identifying high-confidence single- and double-stranded regions. The algorithm was applied to the long non-coding RNA Xist (17 900 nt, not present in the training) and shows an Area under the ROC curve of 0.83 in predicting structured regions.
CROSSalive webserver is freely accessible at http://service.tartaglialab.com/new_submission/crossalive
Supplementary data are available at Bioinformatics online.
1 Introduction
The in vitro structure of an RNA differs from that in vivo for the action of molecules such as RNA-binding proteins (Livi et al., 2015). The complex mechanisms contributing to the formation of structure in vivo are poorly characterized and previous analysis suggests a prevalence of single-stranded regions for all RNA types (Rouskin et al., 2014), although conservation of double-stranded regions has been observed for specific non-coding RNAs (Spitale et al., 2015). In the cellular environment RNA undergoes a number of modifications such as methylation that influence both stability and turnover of the whole transcriptome (Liu and Jia, 2014). Mettl3 is a key component of the complex that methylates adenosine residues at the N6 (m6a) and plays a central role in determining RNA structure in vivo. Indeed, a method of probing RNA structure using the chemical probe NAI-N3 (icSHAPE) indicated that m6a promotes transition from double- to single-stranded regions (Spitale et al., 2015). Through analysis of icSHAPE data we developed the CROSSalive method for the prediction of RNA secondary structure in vivo. One important part of our approach is the use of catRAPID predictions of protein interactions to classify single- and double-stranded regions of RNA molecules (Bellucci et al., 2011). catRAPID estimates the binding through van der Waals, hydrogen bonding and secondary structure properties of both protein and RNA sequences.
2 Workflow and implementation
CROSSalive profiles a RNA sequence computing the corresponding secondary structure in vivo with (m6a+) and without (m6a-) methylation, which is significantly different from that in vitro (Supplementary Fig. S1). The algorithm uses predictions of protein interactions to identify single- and double-stranded regions (Spitale et al., 2015):
For the training and testing we selected RNA fragments carrying the central nucleotide with the highest (single-stranded; 105 non-redundant sequences) and lowest icSHAPE reactivities (double-stranded; 105 non-redundant sequences), following the analysis carried out for CROSS in vitro (Delli Ponti et al., 2017). Each RNA fragment contains a total of 51 nucleotides to allow calculations with catRAPID (Bellucci et al., 2011). The nucleotides are represented as A = (1, 0, 0, 0), C = (0, 1, 0, 0), G = (0, 0, 1, 0) and U = (0, 0, 0, 1).
The catRAPID approach uses a phenomenological potential that exploits several physico-chemical predictors including RNAfold for the RNA structure (Bellucci et al., 2011). 7797 regions from a library of 640 canonical RNA-binding proteins (Agostini et al., 2013) were analyzed to identify those able to discriminate nucleotides in single- and double-stranded states with accuracies >0.6 (m6a+: 228 regions; m6a-: 206 regions; Supplementary Figs S2 and S3).
The dataset is enriched for proteins with gene ontology (Klus et al., 2015) related to RNA structure (double- and single-stranded RNA binding; helicase activity; m6a+: 101 regions; m6a-: 81 regions; Supplementary Tables S1 and S2). The Youden cut-off was computed on catRAPID scores for each protein in the dataset. Scores above the cut-off were set to 1 (0 otherwise).
Neural networks (m6a+ and m6a-, with and without protein contributions) were trained using the architecture described in our previous publication for icSHAPE in vitro (Delli Ponti et al., 2017). Each RNA fragment is assigned a score between -1 (high propensity to be single-stranded) to 1 (high propensity to be double-stranded; Supplementary Fig. S4).
3 Performances
CROSSalive scores were ranked by their absolute value and equal groups of positives and negatives were selected to assess the performances of the algorithm. From low (50%) to high-confidence (HC) scores (1%, Fig. 1A) the accuracy of the models increases monotonically reaching a maximum of 0.86 for the m6a+ model when protein interactions are used (10-fold cross-validation, CV). In comparison, the in vitro icSHAPE model based on RNA sequence information only (Delli Ponti et al., 2017) discriminates single- and double-stranded regions with a 0.88 accuracy (10-fold CV on 1% HC scores). The m6a- in vivo model shows lower accuracy (0.74 in 10-fold CV on 1% HC scores) mainly because m6a removal affects the quality of the training set by altering the stability and turnover of the transcriptome (Liu and Jia, 2014). We applied CROSSalive to an independent in vivo SHAPE-Map experiment (Smola et al., 2016) on the long non-coding Xist (17 900 nt; not in the training). We used the in vivo m6a- model because Mettl3 is poorly abundant in the trophoblasts (Thul et al., 2017) employed in SHAPE-Map and only few nucleotides are methylated at the 5′ and 3′ of Xist (Patil et al., 2016). The algorithm achieves an Area under the ROC curve (AUC) of 0.83 on the 15% HC single- and double-stranded regions ranked by SHAPE reactivity (Fig. 1B). Moreover, CROSSalive profile shows a correlation of 0.45 with the SHAPE-Map one (Fig. 1B). The m6a- model trained on RNA sequence information only achieves an AUC of 0.53 (∼0 correlation).
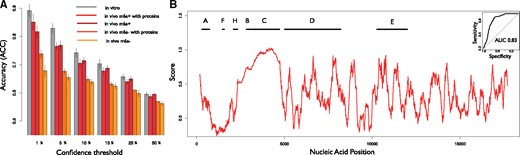
CROSSalive performances. (A) 10-fold cross validation for each specific algorithm (in vitro, in vivo m6a+, in vivo m6a-) with the same training and testing conditions (balanced training set, filtering out sequence redundancy). The accuracies are reported for the scores ranked by their absolute value (same number of positives and negatives were selected), where 50% is the complete set (median). Integrating predictions with protein interactions improves the accuracy. (B) Secondary structure profile of Xist using m6a- model. Known repetitive regions of Xist such as Rep A and Rep C are reported to be very structured (i.e. score > 0). The predicted profile has an overall correlation of 0.45 with in vivo SHAPE data. In the top right we report the ROC curve of CROSSalive on the top and bottom 15% ranked SHAPE data (AUC of 0.83)
4 Conclusions
By using sequence-based information, CROSSalive profiles the RNA secondary structure in vivo. The use of different models (in vivo/in vitro, m6a+/m6a-) will help to identify structural regions to investigate experimentally. As previously done with CROSS (Delli Ponti et al., 2017), CROSSalive can be integrated as a constrain in thermodynamics-based approaches such as RNAfold, which will allow study structural differences of RNAs in vivo and in vitro (Lorenz et al., 2016).
Acknowledgements
The authors thank Andrea Cerase and Alessio Colantoni.
Funding
The research leading to these results has received funding from European Research Council RIBOMYLOME_309545, European Union's Horizon 2020 IASIS_727658 and INFORE_825070, as well as Spanish Ministry of Economy and Competitiveness BFU2017-86970-P.
Conflict of Interest: none declared.
References


{kind=link}