





Abstract
Oxford Nanopore sequencing producing long reads at low cost has made many breakthroughs in genomics studies. However, the large number of errors in Nanopore genome assembly affect the accuracy of genome analysis. Polishing is a procedure to correct the errors in genome assembly and can improve the reliability of the downstream analysis. However, the performances of the existing polishing methods are still not satisfactory.
We developed a novel polishing method, NeuralPolish, to correct the errors in assemblies based on alignment matrix construction and orthogonal Bi-GRU networks. In this method, we designed an alignment feature matrix for representing read-to-assembly alignment. Each row of the matrix represents a read, and each column represents the aligned bases at each position of the contig. In the network architecture, a bi-directional GRU network is used to extract the sequence information inside each read by processing the alignment matrix row by row. After that, the feature matrix is processed by another bi-directional GRU network column by column to calculate the probability distribution. Finally, a CTC decoder generates a polished sequence with a greedy algorithm. We used five real datasets and three assembly tools including Wtdbg2, Flye and Canu for testing, and compared the results of different polishing methods including NeuralPolish, Racon, MarginPolish, HELEN and Medaka. Comprehensive experiments demonstrate that NeuralPolish achieves more accurate assembly with fewer errors than other polishing methods and can improve the accuracy of assembly obtained by different assemblers.
Supplementary data are available at Bioinformatics online.
1 Introduction
With the development of Oxford Nanopore Technologies (ONT), Nanopore long-read sequencing has made significant progress in the fields of genomics (Jain et al., 2018), transcriptomics (Garalde et al., 2018) and epigenomics (Ni et al., 2019). Compared with second-generation sequencing technologies, Nanopore sequencing has the advantages of long reads, no PCR amplification, no GC bias and real-time analysis. When the long reads span repetitive elements, repeat copies can be anchored within the genome’s unique position. Thus such long-read-based sequencing can provide great potentials for de novo genome assembly.
MinION is a portable long-read sequencing instrument from Oxford Nanopore Technologies that enables direct, real-time analysis of long DNA or RNA fragments, which can produce reads longer than 800 kb (Jain et al., 2018). It works by measuring electrical current changes as single stranded DNA/RNA segments passing through a protein nanopore, and the raw signals are translated into DNA or RNA sequences by basecalling tools. The main limitation of Nanopore sequencing is its high error rate compared to the short-read sequencing technology. With the development of nanopore chemistry and basecaller, the error rate of reads still ranges from 5% to 20%. The error-prone reads usually lead to a highly erroneous assembly, quite different from the subject’s actual genome, thus causing the downstream genome analysis task results to be unreliable.
Currently, there are two main ways of reducing errors in Nanopore genome assemblies. The first way is to correct the reads used for genome assembly. Most errors in the reads are random so that they can be corrected with high coverage. FC_Consensus (Chin et al., 2016), DAGCon (Chin et al., 2013) and FalconSense (Berlin et al., 2015) are the three most widely used long read error correction methods embedded in assemblers. Chin et al. (2013) presented a hierarchical genome assembly process (HGAP) for high-quality de novo microbial genome assemblies. In this process, they developed the correction method named DAGCon to improve the quality of raw reads for assembly. DAGCon represents a multiple sequence alignment of a read that needs to be corrected as a partial order graph (POG) (Lee et al., 2002) and uses a dynamic programming algorithm to find the corrected sequence. Koren et al. (2017) developed the long reads assembler named Canu as a successor of Celera Assembler. In Canu, the raw reads used for genome assembly are corrected using FC_Consensus, which is also an error correction algorithm by constructing a POG from all-versus-all read alignment. In MECAT, Xiao et al. (2017) combined the principles from both DAGCon and FalconSense. FalconSense corrects the reads by counting consistent bases in the alignment. For regions with consistent matches/deletions without insertions (simple regions), they applied FalconSense. Moreover, for other complicate regions, they used the modified DAGCon. Even though these assemblers correct the raw reads before assembly, users still need to polish the assembly with additional polishing tools if a more accurate assembly is required.
The second way to reduce the errors in Nanopore genome assembly and improve the assembly quality is polishing. The polishing algorithms usually take the read-to-assembly alignment as the input, then compare the draft assembly to the mapping reads and determine if the assembly’s bases need to be modified. Currently, there are three main strategies for polishing. The first polishing strategy uses short reads to correct the assembly generated from long reads. Pilon (Walker et al., 2014), ntEdit (Warren et al., 2019), NextPolish (Hu et al., 2020), Apollo (Firtina et al., 2020) and POLCA (Zimin and Salzberg, 2020) are representative tools of this kind. These polishing tools align highly accurate short reads to the assembly and correct the assembly from reads alignment. The second strategy is to use additional sequencing information. Since the raw signals recorded by the Nanopore sequencer contain more information than basecalled reads, Nanopolish (Loman et al., 2015) tries to use these currents to reduce errors in genome assembly. Nanopolish calculates the probability of each raw signal sequence based on a profile hidden Markov model (HMM). The core algorithm of Nanopolish is to apply signal-to-assembly alignment to modify the draft assembly, and then find a modified sequence that maximizes the sum of all current emission probabilities. The third strategy is to polish the assembly with long reads. Racon, Medaka, MarginPolish and HELEN are all based on this idea. Racon (Vaser et al., 2017) is a highly efficient consensus module based on the POG. The read-to-assembly alignment of the draft assembly is used to construct a POG. According to the graph, the final consensus sequence is calculated by a dynamic programming algorithm. Medaka (https://github.com/nanoporetech/medaka), developed by Oxford Nanopore Technologies, is a tool to create a consensus sequence from Nanopore sequencing data. To our knowledge, it is the first polishing tool based on neural networks. Medaka counts the occurrences of different bases at each position on the assembly. A recurrent neural network is used to capture the correlation of base counts on different positions of the assembly and to predict the true base at each position. From the reports on the official website (https://nanoporetech.github.io/medaka/benchmarks.html), it improves accuracy over graph-based methods. It can compete with the state-of-the-art signal-based approach, Nanopolish, with a much higher speed. Shafin et al. (2020) developed a polishing pipeline named ‘MarginPolish & HELEN’ based on a deep neural network. It is designed to improve the base-level quality of the draft assembly. There are two modules in this pipeline, MarginPolish and HELEN. MarginPolish uses a pairwise hidden Markov model (pair-HMM) to produce pairwise alignment statistics from read-to-assembly alignments. HELEN comprises a multi-task recurrent neural network (RNN) that utilizes the weights of the POG in MarginPolish. HELEN finally predicts a nucleotide base and the number of repetitions of this base for each genomic position.
Despite the advantages in Nanopore polishing algorithms, the polishing methods based on the second-generation short read require extra sequencing reads from other sequencing platforms, which increases the cost of sequencing. Since the length of the raw signal is close to 8–9 times of the read length, the time cost of the polishing methods based on the raw signal is unbearable. Thus the strategy for polishing based on long reads is more promising. However, there are still some limitations for existing methods. Racon can calculate the optimal path from the POG, but the path with the maximum score based on the Racon scoring model does not necessarily represent the true consensus sequence. Medaka considers the context information of alignment by the base distribution at each position of the contig but loses the context information inside each read.
Here, we present a novel polishing method, named NeuralPolish, based on alignment matrix construction and orthogonal Bi-GRU networks. NeuralPolish only uses basecalled reads to correct the assembly’s errors by aligning reads to the draft assembly. To feed the read-to-assembly alignment into a neural network for prediction, we construct an alignment matrix from the read-to-assembly alignment. In the network architecture, we pair two bi-directional GRU networks, orthogonally, with a CTC decoder (Graves et al., 2006) to calculate the polished contigs. We compare the accuracy of different polishing tools including NeuralPolish, Racon, Medaka, MarginPolish and HELEN on five real datasets. To evaluate the performance of polishing tools on the assembly generated by different assemblers, we use three different assemblers including Wtdbg2, Flye and Canu to generate the draft assembly of each dataset. NeuralPolish can achieve polished results with fewer errors in these tests compared to Racon, Medaka, MarginPolish and HELEN.
2 Materials and methods
In this section, we will describe the new polishing approach named NeuralPolish. It corrects genome assemblies with Nanopore basecalled reads relying on a recurrent neural network. The contents are organized as follows. First, we will introduce the datasets of Nanopore reads used in our experiment and the feature construction from read-to-assembly alignment. Then we will describe the pipeline in NeuralPolish. In the end, we will introduce the architecture of the neural network in NeuralPolish.
2.1 Datasets
Our experiment uses five real datasets including Escherichia coli K12, Saccharomyces cerevisiae w303, Homo sapiens (NA12878) chromosome 21, Homo sapiens (NA12878) chromosome 20 and Arabidopsis thaliana. All of the raw data and references are available from public websites as shown in Supplementary Table S1. Here, the two human chromosomes are processed separately because part of the NA12878 chromosome 21 is used to train NeuralPolish and the NA12878 chromosome 20 is never seen by NeuralPolish during evaluation.
2.2 Alignment matrix construction
In several polishing tools, the read-to-assembly alignments are preprocessed into the algorithm’s input data in different ways. Racon constructs a POG from multiple sequence alignment of the contig to be polished. Medaka counts the number of different bases at each position of the contig from the read-to-assembly alignment. In NeuralPolish, we propose to construct a novel alignment matrix from the read-to-assembly alignment. The construction of the alignment matrix is shown in Figure 1. Each read is aligned to the contig by long reads aligner. We record the entire information of alignment between each read and draft assembly in the alignment matrix. When there is an insertion in the alignment, a nucleotide base in the contig responds to multiple nucleotide bases in the read. The insertion bases are combined with its left-hand side base, which is consistent with SAMtools (Li et al., 2009). Once an insertion occurs, we record the k-mer with multiple bases to capture the insertion, so that it is clear where the insertion is and what the insertion bases are in the alignment matrix. For a deletion, there is no base aligned to the contig, so we use a symbol ‘-’ to represent the missing of the base. At the positions outside the range of read alignment, there are also no bases aligned to the contig, so we use the symbol ‘*’ to fill the blank. The number of columns in the matrix equals to the size of the window to be polished, and the number of rows in the matrix equals to the coverage of reads. In the matrix, each row represents a read and each column represents the aligned bases at each position of the contig. Each element in the matrix represents the aligned base, the insertion bases or the deletion. For the sake of computational efficiency, we encode the element in the matrix as integers. The deletion symbol ‘-’ is encoded with the integer 0, and the padding symbol ‘*’ is encoded with the integer 1. Nucleotide characters A, T, C and G are encoded as integers 2–5. All 2-mers are encoded as integers 6–21. All 3-mers are encoded as integers 22–85. All 4-mers are encoded as integers 86–341. All 5-mers are encoded as integers 342–1365. Here, each element of the matrix is up to 5-mer. The reason is that, to learn the language relationship between different k-mers, there is an embedding layer in the neural network to process the input data. If each element of the matrix can be encoded into a longer k-mer, the number of words and the space of embedding layer will grow exponentially, which will affect computational efficiency.
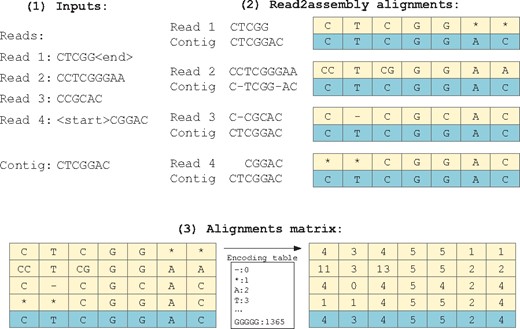
The construction of the alignment matrix from read-to-assembly alignment. In the alignment matrix, the blue row is the contig sequence and yellow rows are aligned reads. The asterisk indicates that there is no bases aligned to the contig. The character ‘-’ represents a deletion
2.3 Pipeline in NeuralPolish
As shown in Figure 2A, the polishing pipeline in NeuralPolish contains four steps. The input data of the pipeline is the draft assembly and raw reads. The draft assembly can be produced by different long-read assemblers such as Canu (Koren et al., 2017), Flye (Kolmogorov et al., 2019), Wtdbg2 (Ruan and Li, 2020) and Necat (Chen et al., 2021). In the workflow of NeuralPolish, we process the draft assembly with one round of Racon. This is because the two state-of-the-art Nanopore assembly polishing workflows, Medaka and MarginPolish & HELEN, use a similar framework, a graph-based polishing followed by a neural network-based polishing. The polishing procedure of Medaka consists of four rounds of Racon and one round of Medaka. MarginPolish & HELEN polishing is made up of one round of MarginPolish and one round of HELEN. Graph-based polishing method can help to solve long-length structural errors in the assembly, while the neural network-based approach mainly solves fine base-level errors including substitution and small indels. Both types of the errors are important, and solving only one of them will not achieve best polishing accuracy. We evaluate the performance of different rounds of Racon in the NeuralPolish workflow and the error rate of polishing results is shown in Supplementary Figure S1. NeuralPolish with one round of Racon can significantly reduce the error rate of the polishing result than NeuralPolish without Racon. More rounds of Racon in NeuralPolish can further reduce the error rate, but they also bring more computational expense. Then the raw reads are aligned to the polished assembly produced by Racon with minimap2 (Li, 2018). After that, the read-to-assembly alignments are used to construct an alignment matrix as the neural network’s input data, as described in Section 2.2. Finally, the trained recurrent neural network predicts the polished sequence.
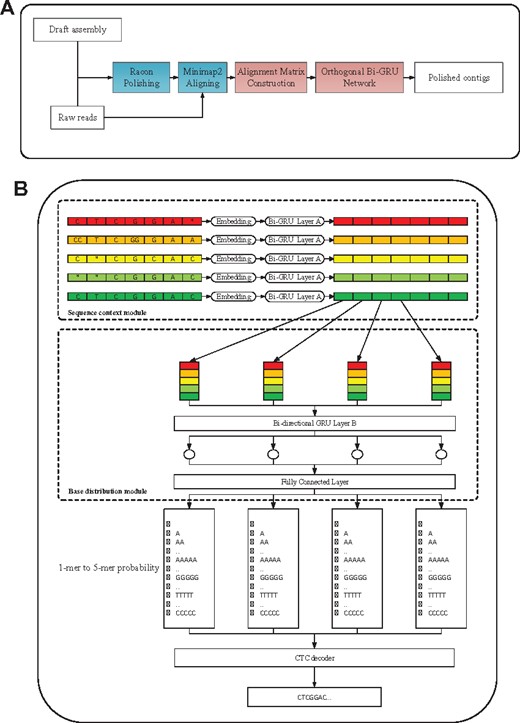
(A) Overview of the polishing pipeline in NeuralPolish. The first step in NeuralPolish is to polish the draft assembly with one round of Racon. Then the raw reads are aligned to the polished assembly generated by Racon. Next, NeuralPolish constructs an alignment matrix from the read-to-assembly alignment. Finally, the trained orthogonal bi-directional GRU network predicts the polished contigs from the alignment matrix. (B) Architecture of neural network in NeuralPolish. The network is mainly composed of two orthogonal bi-directional GRU network and a CTC decoder. The bi-directional GRU layer A is used to process the alignment matrix by rows and the bi-directional GRU layer B processes the feature matrix by columns. The CTC decoder is used to generate the final polished contig
2.4 Network architecture
In this section, we will describe the architecture of the NeuralPolish network. As shown in Figure 2B, NeuralPolish comprises three modules including the sequence context module, the base distribution module and the CTC decoder module. The sequence context module learns the sequence context information in each aligned read. The base distribution module is mainly responsible for calculating the base distribution of each position. Thus these two modules incorporate orthogonal information in the alignment matrix. The CTC decoder module is to generate the most likely polished sequence with the greedy search algorithm.
2.4.1 Sequence context module
We train a bi-directional RNN (BRNN) network in the sequence context module to extract sequence context features from each aligned read. The BRNN contains a forward RNN and a backward RNN to capture both upstream and downstream sequence information. Since Gated Recurrent Unit (GRU) uses less training parameters and executes faster than long short-term memory (LSTM) (Chung et al., 2014), we use GRU in each RNN. In the sequence context module, each row in the alignment matrix is processed individually by the Bi-GRU network. Each read is computed separately to avoid being affected by other reads when calculating the sequence information of the current read. In the last part of the sequence context module, the sequence context information of multiple reads in the alignment matrix is stacked into a column.
2.4.2 Base distribution module
In the base distribution module, we pair a Bi-GRU network and a fully connected layer to calculate the base distribution at each contig position. The GRU network scans the stacked sequence context information at each contig position to calculate the hidden state. To enable the model to polish deletion errors, the fully connected layer is used to convert the output of the GRU layer at each position of the contig to the probability of k-mers () or the blank symbol. Since each position of the contig can be predicted as a 5-mer at most, an n-mer in the contig can be converted to at most bases in the polished result. This strategy ensures that NeuralPolish can handle very long indel errors.
2.4.3 CTC decoder module
In the CTC decoder module, we use Connectionist Temporal Classification (CTC) (Graves et al., 2006), an algorithm used to train deep neural networks in sequence problems. CTC is a sequence prediction method without knowing the alignment between input and output. The CTC decoder module is attached to the base distribution module and takes the probability of k-mers from the previous layer as input. Let be the sequence of k-mers probability on m positions, and be the corresponding target DNA sequence. There are several challenges when training a neural network with other supervised learning algorithms. (i) The length of input sequence X may vary from the length of target sequence Y. (ii) The ratio of the length between X and Y is not fixed. (iii) We do not know the accurate element-wise alignment of input sequence X and target sequence Y. Fortunately, the CTC algorithm overcomes these challenges. For a give input sequence X, the CTC algorithm provides all possible output distributions . We can use these distributions to infer the possible output sequence or evaluate the probability of a given output sequence. During testing, the CTC decoder uses a greedy search algorithm to pick the symbol with the largest probability in each contig position. By removing the blank symbols and chaining the remaining symbols together to form a sequence, the CTC decoder ultimately provides the polished sequence.
2.4.4 Parameters and training strategy
There are some hyper-parameters in the NeuralPolish network. The Bi-GRU network in the sequence context module consists of three bi-directional GRU layers and the hidden size of each layer is set to 32. The Bi-GRU network in the base distribution module comprises three bi-directional GRU layers and the hidden size of each layer is set to 256. The batch size used during model training is 128. The Adam optimizer (Kingma and Ba, 2014) is used to optimize the CTC loss function and the learning rate is 0.001. We use gradient clipping to clip the excessive gradient value to a threshold value to prevent gradient explosion during training. The gradient clipping threshold is set to 1. To prevent over-fitting, we use the dropout strategy to randomly ignore the values of some neural cells. The dropout ratio is set to 0.5.
3 Experiments and results
3.1 Training and test datasets
To train the NeuralPolish model, we constructed a training dataset with three genomes including E.coli, S.cerevisiae and NA12878 chromosome 21. Firstly, we used Wtdbg2 to assemble the raw reads and polished the draft assemblies with one round of Racon. Afterwards, the raw reads are aligned to the Racon polished assemblies by minimap2 to generate the read-to-assembly alignments. The reference of each genome is also aligned to the Racon polished assemblies to generate the ground-truth labellings. Since NeuralPolish polishes a 64 bp window (without overlaps) of the contig each time, we used a window with the same length to randomly pick some fragments from the contigs. The window size of 64 is determined according to the memory of the GPU device. We would like to clarify that the setting of the window size will not affect the accuracy of polishing results. Then we constructed alignment matrices of these fragments for training the model. The training dataset is mixed with 10 000 fragments from the assembly of E.coli (14% of the E.coli genome), 10 000 fragments from the assembly of S.cerevisiae (5% of the S.cerevisiae genome) and 20 000 fragments from the assembly of NA12878 chromosome 21 (3% of the NA12878 chromosome 21 genome). The training phase of the NeuralPolish model on a Nvidia GeForce RTX 2080Ti took 20 hours. Besides the training dataset, the test dataset was made up of five genomes including E.coli, S.cerevisiae, NA12878 chromosome 21, NA12878 chromosome 20 and A.thaliana. The test dataset used all window fragments except the ones included in the training. Since the assembly results can be generated by different assemblers, we used Flye, Wtdbg2 and Canu to produce assembly of each genome.
Pomoxis (https://github.com/nanoporetech/pomoxis) is a bioinformatics tool developed by Oxford Nanopore Technologies (ONT), which was used as the accuracy assessment tool to compare the performance of Medaka and Nanopolish by ONT researchers. Pomoxis reports a mismatch error rate, an insertion error rate and a deletion error rate. The mismatch, deletion and insertion rates are defined as the number of mismatched, deleted and inserted bases divided by the number of bases in the reference sequence. The total error rate is defined as the sum of mismatch, deletion and insertion error rates. Here, we used Pomoxis to compare the error rates of the assemblies processed by different polishing methods.
3.2 Polishing assemblies generated by Wtdbg2, Flye and Canu
Wtdbg2 is a de novo sequence assembler based on the fuzzy Bruijn graph (Ruan and Li, 2020). To test the performance of different polishing methods on the assembly of Wtdbg2, each genome in the test dataset was assembled by Wtdbg2 (v2.5) and polished by Racon, Medaka, MarginPolish, HELEN and NeuralPolish, respectively. For Racon polishing, we performed four rounds of Racon then obtained the final polished assemblies. According to the usage of Medaka recommended by ONT, the draft assemblies were processed with four rounds of Racon and one round of Medaka. For MarginPolish and HELEN, each draft assembly was polished with one round of MarginPolish and one round of HELEN. For NeuralPolish, the draft assemblies were processed with one round of Racon and one round of NeuralPolish. Here all the assembly, alignment and polishing process used the default parameters for Nanopore reads.
When polishing the draft assemblies produced by Wtdbg2, the error rates of different polishing methods are presented in Table 1. NeuralPolish achieves the lowest error rate on four out of the five test datasets. On the data of NA12878 chromosome 21 and 20, NeuralPolish has the most obvious improvement in the error rate of polishing results compared to other tools. The polishing tool with the second lowest error rate is Medaka, while NeuralPolish has an improvement rate of 26.6% and 15% over Medaka on these two datasets. This is because NeuralPolish resolves a large number of deletion errors. On the data of S.cerevisiae, the error rate of contigs polished by NeuralPolish is not as low as that of contigs polished by Medaka, MarginPolish and HELEN. The reason is that NeuralPolish is more powerful in resolving deletion errors, but there are not many deletion errors in the contigs of S.cerevisiae. Therefore NeuralPolish plays a limited role in improving the accuracy of this dataset.
Comparison of the error rates of polishing results by Racon, Medaka, MarginPolish, HELEN and NeuralPolish on the data assembled by Wtdbg2. The bold in the table means the best results.
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Wtdbg2 | 0.07% | 0.32% | 0.03% | 0.42% |
| Racon | 0.11% | 0.18% | 0.13% | 0.42% | |
| Medaka | 0.13% | 0.05% | 0.10% | 0.28% | |
| MarginPolish | 0.10% | 0.09% | 0.10% | 0.29% | |
| HELEN | 0.09% | 0.13% | 0.10% | 0.31% | |
| NeuralPolish | 0.06% | 0.11% | 0.07% | 0.24% | |
| S.cerevisiae | Wtdbg2 | 0.11% | 1.08% | 0.53% | 1.72% |
| Racon | 0.11% | 0.77% | 0.67% | 1.55% | |
| Medaka | 0.09% | 0.57% | 0.55% | 1.21% | |
| MarginPolish | 0.08% | 0.59% | 0.54% | 1.21% | |
| HELEN | 0.09% | 0.73% | 0.58% | 1.39% | |
| NeuralPolish | 0.10% | 0.67% | 0.74% | 1.51% | |
| NA12878 chr21 | Wtdbg2 | 0.26% | 4.84% | 0.24% | 5.34% |
| Racon | 0.26% | 2.82% | 0.31% | 3.39% | |
| Medaka | 0.32% | 2.47% | 0.29% | 3.08% | |
| MarginPolish | 0.28% | 2.71% | 0.31% | 3.31% | |
| HELEN | 1.50% | 9.02% | 0.87% | 11.38% | |
| NeuralPolish | 0.28% | 1.50% | 0.48% | 2.26% | |
| NA12878 chr20 | Wtdbg2 | 0.27% | 5.20% | 0.21% | 5.68% |
| Racon | 0.27% | 3.04% | 0.32% | 3.63% | |
| Medaka | 0.33% | 2.66% | 0.27% | 3.26% | |
| MarginPolish | 0.43% | 3.26% | 0.35% | 4.04% | |
| HELEN | 1.76% | 9.60% | 0.95% | 12.31% | |
| NeuralPolish | 0.39% | 1.87% | 0.51% | 2.77% | |
| A.thaliana | Wtdbg2 | 0.69% | 5.45% | 1.85% | 7.99% |
| Racon | 0.68% | 3.70% | 1.89% | 6.27% | |
| Medaka | 0.68% | 3.42% | 2.01% | 6.11% | |
| MarginPolish | 0.70% | 4.15% | 2.02% | 6.87% | |
| HELEN | 1.73% | 6.86% | 2.28% | 10.87% | |
| NeuralPolish | 0.69% | 3.05% | 2.26% | 6.00% |
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Wtdbg2 | 0.07% | 0.32% | 0.03% | 0.42% |
| Racon | 0.11% | 0.18% | 0.13% | 0.42% | |
| Medaka | 0.13% | 0.05% | 0.10% | 0.28% | |
| MarginPolish | 0.10% | 0.09% | 0.10% | 0.29% | |
| HELEN | 0.09% | 0.13% | 0.10% | 0.31% | |
| NeuralPolish | 0.06% | 0.11% | 0.07% | 0.24% | |
| S.cerevisiae | Wtdbg2 | 0.11% | 1.08% | 0.53% | 1.72% |
| Racon | 0.11% | 0.77% | 0.67% | 1.55% | |
| Medaka | 0.09% | 0.57% | 0.55% | 1.21% | |
| MarginPolish | 0.08% | 0.59% | 0.54% | 1.21% | |
| HELEN | 0.09% | 0.73% | 0.58% | 1.39% | |
| NeuralPolish | 0.10% | 0.67% | 0.74% | 1.51% | |
| NA12878 chr21 | Wtdbg2 | 0.26% | 4.84% | 0.24% | 5.34% |
| Racon | 0.26% | 2.82% | 0.31% | 3.39% | |
| Medaka | 0.32% | 2.47% | 0.29% | 3.08% | |
| MarginPolish | 0.28% | 2.71% | 0.31% | 3.31% | |
| HELEN | 1.50% | 9.02% | 0.87% | 11.38% | |
| NeuralPolish | 0.28% | 1.50% | 0.48% | 2.26% | |
| NA12878 chr20 | Wtdbg2 | 0.27% | 5.20% | 0.21% | 5.68% |
| Racon | 0.27% | 3.04% | 0.32% | 3.63% | |
| Medaka | 0.33% | 2.66% | 0.27% | 3.26% | |
| MarginPolish | 0.43% | 3.26% | 0.35% | 4.04% | |
| HELEN | 1.76% | 9.60% | 0.95% | 12.31% | |
| NeuralPolish | 0.39% | 1.87% | 0.51% | 2.77% | |
| A.thaliana | Wtdbg2 | 0.69% | 5.45% | 1.85% | 7.99% |
| Racon | 0.68% | 3.70% | 1.89% | 6.27% | |
| Medaka | 0.68% | 3.42% | 2.01% | 6.11% | |
| MarginPolish | 0.70% | 4.15% | 2.02% | 6.87% | |
| HELEN | 1.73% | 6.86% | 2.28% | 10.87% | |
| NeuralPolish | 0.69% | 3.05% | 2.26% | 6.00% |
Comparison of the error rates of polishing results by Racon, Medaka, MarginPolish, HELEN and NeuralPolish on the data assembled by Wtdbg2. The bold in the table means the best results.
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Wtdbg2 | 0.07% | 0.32% | 0.03% | 0.42% |
| Racon | 0.11% | 0.18% | 0.13% | 0.42% | |
| Medaka | 0.13% | 0.05% | 0.10% | 0.28% | |
| MarginPolish | 0.10% | 0.09% | 0.10% | 0.29% | |
| HELEN | 0.09% | 0.13% | 0.10% | 0.31% | |
| NeuralPolish | 0.06% | 0.11% | 0.07% | 0.24% | |
| S.cerevisiae | Wtdbg2 | 0.11% | 1.08% | 0.53% | 1.72% |
| Racon | 0.11% | 0.77% | 0.67% | 1.55% | |
| Medaka | 0.09% | 0.57% | 0.55% | 1.21% | |
| MarginPolish | 0.08% | 0.59% | 0.54% | 1.21% | |
| HELEN | 0.09% | 0.73% | 0.58% | 1.39% | |
| NeuralPolish | 0.10% | 0.67% | 0.74% | 1.51% | |
| NA12878 chr21 | Wtdbg2 | 0.26% | 4.84% | 0.24% | 5.34% |
| Racon | 0.26% | 2.82% | 0.31% | 3.39% | |
| Medaka | 0.32% | 2.47% | 0.29% | 3.08% | |
| MarginPolish | 0.28% | 2.71% | 0.31% | 3.31% | |
| HELEN | 1.50% | 9.02% | 0.87% | 11.38% | |
| NeuralPolish | 0.28% | 1.50% | 0.48% | 2.26% | |
| NA12878 chr20 | Wtdbg2 | 0.27% | 5.20% | 0.21% | 5.68% |
| Racon | 0.27% | 3.04% | 0.32% | 3.63% | |
| Medaka | 0.33% | 2.66% | 0.27% | 3.26% | |
| MarginPolish | 0.43% | 3.26% | 0.35% | 4.04% | |
| HELEN | 1.76% | 9.60% | 0.95% | 12.31% | |
| NeuralPolish | 0.39% | 1.87% | 0.51% | 2.77% | |
| A.thaliana | Wtdbg2 | 0.69% | 5.45% | 1.85% | 7.99% |
| Racon | 0.68% | 3.70% | 1.89% | 6.27% | |
| Medaka | 0.68% | 3.42% | 2.01% | 6.11% | |
| MarginPolish | 0.70% | 4.15% | 2.02% | 6.87% | |
| HELEN | 1.73% | 6.86% | 2.28% | 10.87% | |
| NeuralPolish | 0.69% | 3.05% | 2.26% | 6.00% |
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Wtdbg2 | 0.07% | 0.32% | 0.03% | 0.42% |
| Racon | 0.11% | 0.18% | 0.13% | 0.42% | |
| Medaka | 0.13% | 0.05% | 0.10% | 0.28% | |
| MarginPolish | 0.10% | 0.09% | 0.10% | 0.29% | |
| HELEN | 0.09% | 0.13% | 0.10% | 0.31% | |
| NeuralPolish | 0.06% | 0.11% | 0.07% | 0.24% | |
| S.cerevisiae | Wtdbg2 | 0.11% | 1.08% | 0.53% | 1.72% |
| Racon | 0.11% | 0.77% | 0.67% | 1.55% | |
| Medaka | 0.09% | 0.57% | 0.55% | 1.21% | |
| MarginPolish | 0.08% | 0.59% | 0.54% | 1.21% | |
| HELEN | 0.09% | 0.73% | 0.58% | 1.39% | |
| NeuralPolish | 0.10% | 0.67% | 0.74% | 1.51% | |
| NA12878 chr21 | Wtdbg2 | 0.26% | 4.84% | 0.24% | 5.34% |
| Racon | 0.26% | 2.82% | 0.31% | 3.39% | |
| Medaka | 0.32% | 2.47% | 0.29% | 3.08% | |
| MarginPolish | 0.28% | 2.71% | 0.31% | 3.31% | |
| HELEN | 1.50% | 9.02% | 0.87% | 11.38% | |
| NeuralPolish | 0.28% | 1.50% | 0.48% | 2.26% | |
| NA12878 chr20 | Wtdbg2 | 0.27% | 5.20% | 0.21% | 5.68% |
| Racon | 0.27% | 3.04% | 0.32% | 3.63% | |
| Medaka | 0.33% | 2.66% | 0.27% | 3.26% | |
| MarginPolish | 0.43% | 3.26% | 0.35% | 4.04% | |
| HELEN | 1.76% | 9.60% | 0.95% | 12.31% | |
| NeuralPolish | 0.39% | 1.87% | 0.51% | 2.77% | |
| A.thaliana | Wtdbg2 | 0.69% | 5.45% | 1.85% | 7.99% |
| Racon | 0.68% | 3.70% | 1.89% | 6.27% | |
| Medaka | 0.68% | 3.42% | 2.01% | 6.11% | |
| MarginPolish | 0.70% | 4.15% | 2.02% | 6.87% | |
| HELEN | 1.73% | 6.86% | 2.28% | 10.87% | |
| NeuralPolish | 0.69% | 3.05% | 2.26% | 6.00% |
To test whether NeuralPolish can be applied to polishing the draft assemblies produced by other assemblers, we used the NeuralPolish model trained on Wtdbg2 assemblies to polish Flye assemblies and Canu assemblies, without re-training. Flye is a de novo assembler for single molecule sequencing reads using repeat graphs (Kolmogorov et al., 2019), and Canu is a branch of the Celera Assembler, designed for high-noise single-molecule sequencing (Koren et al., 2017). The execution process of each polishing method is the same as the process executed during polishing Wtdbg2 assemblies.
We used Flye to assemble the raw reads and polished the contigs with different polishing tools. The error rates of polished contigs by Racon, Medaka, MarginPolish, HELEN and NeuralPolish are shown in Table 2. We can see that although NeuralPolish is trained on Wtdbg2 assemblies, it still achieves the lowest error rates of polished contigs on four out of the five test datasets assembled by Flye. The reduction in the error rate of NeuralPolish results is most significant for the data of NA12878 chromosome 21 and 20. Medaka and MarginPolish have the second lowest error rates on chromosome 21 and chromosome 20, respectively. NeuralPolish has an improvement rate of 20.7% over Medaka on chromosome 21 and an improvement rate of 17.5% over MarginPolish on chromosome 20. In terms of Canu assemblies, we used Canu to assemble the raw reads and polished them with different tools. The error rates of polished results by Racon, Medaka, MarginPolish, HELEN and NeuralPolish are provided in Supplementary Table S2. Consistent with the other experiments, on four out of the five test datasets, NeuralPolish gets the lowest error rate of polished contigs.
Comparison of the error rates of polishing results by Racon, Medaka, MarginPolish, HELEN and NeuralPolish on the data assembled by Flye. The bold in the table means the best results.
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Flye | 0.18% | 0.09% | 0.07% | 0.34% |
| Racon | 0.11% | 0.17% | 0.13% | 0.41% | |
| Medaka | 0.14% | 0.05% | 0.10% | 0.29% | |
| MarginPolish | 0.11% | 0.08% | 0.11% | 0.30% | |
| HELEN | 0.09% | 0.12% | 0.10% | 0.31% | |
| NeuralPolish | 0.07% | 0.10% | 0.08% | 0.25% | |
| S.cerevisiae | Flye | 0.10% | 0.75% | 0.50% | 1.35% |
| Racon | 0.12% | 0.75% | 0.63% | 1.50% | |
| Medaka | 0.10% | 0.54% | 0.52% | 1.16% | |
| MarginPolish | 0.09% | 0.60% | 0.40% | 1.09% | |
| HELEN | 0.09% | 0.74% | 0.44% | 1.26% | |
| NeuralPolish | 0.11% | 0.69% | 0.58% | 1.38% | |
| NA12878 chr21 | Flye | 0.39% | 2.90% | 0.32% | 3.61% |
| Racon | 0.37% | 2.84% | 0.42% | 3.63% | |
| Medaka | 0.42% | 2.38% | 0.38% | 3.19% | |
| MarginPolish | 0.33% | 2.25% | 0.26% | 2.84% | |
| HELEN | 2.09% | 17.30% | 0.48% | 19.88% | |
| NeuralPolish | 0.42% | 1.56% | 0.55% | 2.53% | |
| NA12878 chr20 | Flye | 0.33% | 3.08% | 0.27% | 3.68% |
| Racon | 0.31% | 2.99% | 0.36% | 3.66% | |
| Medaka | 0.35% | 2.50% | 0.32% | 3.16% | |
| MarginPolish | 0.28% | 2.32% | 0.32% | 2.92% | |
| HELEN | 2.11% | 16.83% | 0.59% | 19.53% | |
| NeuralPolish | 0.34% | 1.56% | 0.50% | 2.40% | |
| A.thaliana | Flye | 0.73% | 3.94% | 1.99% | 6.66% |
| Racon | 0.69% | 3.57% | 2.11% | 6.37% | |
| Medaka | 0.74% | 3.47% | 2.03% | 6.23% | |
| MarginPolish | 0.69% | 3.49% | 2.05% | 6.23% | |
| HELEN | 1.07% | 4.79% | 2.11% | 7.97% | |
| NeuralPolish | 0.73% | 2.94% | 2.37% | 6.04% |
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Flye | 0.18% | 0.09% | 0.07% | 0.34% |
| Racon | 0.11% | 0.17% | 0.13% | 0.41% | |
| Medaka | 0.14% | 0.05% | 0.10% | 0.29% | |
| MarginPolish | 0.11% | 0.08% | 0.11% | 0.30% | |
| HELEN | 0.09% | 0.12% | 0.10% | 0.31% | |
| NeuralPolish | 0.07% | 0.10% | 0.08% | 0.25% | |
| S.cerevisiae | Flye | 0.10% | 0.75% | 0.50% | 1.35% |
| Racon | 0.12% | 0.75% | 0.63% | 1.50% | |
| Medaka | 0.10% | 0.54% | 0.52% | 1.16% | |
| MarginPolish | 0.09% | 0.60% | 0.40% | 1.09% | |
| HELEN | 0.09% | 0.74% | 0.44% | 1.26% | |
| NeuralPolish | 0.11% | 0.69% | 0.58% | 1.38% | |
| NA12878 chr21 | Flye | 0.39% | 2.90% | 0.32% | 3.61% |
| Racon | 0.37% | 2.84% | 0.42% | 3.63% | |
| Medaka | 0.42% | 2.38% | 0.38% | 3.19% | |
| MarginPolish | 0.33% | 2.25% | 0.26% | 2.84% | |
| HELEN | 2.09% | 17.30% | 0.48% | 19.88% | |
| NeuralPolish | 0.42% | 1.56% | 0.55% | 2.53% | |
| NA12878 chr20 | Flye | 0.33% | 3.08% | 0.27% | 3.68% |
| Racon | 0.31% | 2.99% | 0.36% | 3.66% | |
| Medaka | 0.35% | 2.50% | 0.32% | 3.16% | |
| MarginPolish | 0.28% | 2.32% | 0.32% | 2.92% | |
| HELEN | 2.11% | 16.83% | 0.59% | 19.53% | |
| NeuralPolish | 0.34% | 1.56% | 0.50% | 2.40% | |
| A.thaliana | Flye | 0.73% | 3.94% | 1.99% | 6.66% |
| Racon | 0.69% | 3.57% | 2.11% | 6.37% | |
| Medaka | 0.74% | 3.47% | 2.03% | 6.23% | |
| MarginPolish | 0.69% | 3.49% | 2.05% | 6.23% | |
| HELEN | 1.07% | 4.79% | 2.11% | 7.97% | |
| NeuralPolish | 0.73% | 2.94% | 2.37% | 6.04% |
Comparison of the error rates of polishing results by Racon, Medaka, MarginPolish, HELEN and NeuralPolish on the data assembled by Flye. The bold in the table means the best results.
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Flye | 0.18% | 0.09% | 0.07% | 0.34% |
| Racon | 0.11% | 0.17% | 0.13% | 0.41% | |
| Medaka | 0.14% | 0.05% | 0.10% | 0.29% | |
| MarginPolish | 0.11% | 0.08% | 0.11% | 0.30% | |
| HELEN | 0.09% | 0.12% | 0.10% | 0.31% | |
| NeuralPolish | 0.07% | 0.10% | 0.08% | 0.25% | |
| S.cerevisiae | Flye | 0.10% | 0.75% | 0.50% | 1.35% |
| Racon | 0.12% | 0.75% | 0.63% | 1.50% | |
| Medaka | 0.10% | 0.54% | 0.52% | 1.16% | |
| MarginPolish | 0.09% | 0.60% | 0.40% | 1.09% | |
| HELEN | 0.09% | 0.74% | 0.44% | 1.26% | |
| NeuralPolish | 0.11% | 0.69% | 0.58% | 1.38% | |
| NA12878 chr21 | Flye | 0.39% | 2.90% | 0.32% | 3.61% |
| Racon | 0.37% | 2.84% | 0.42% | 3.63% | |
| Medaka | 0.42% | 2.38% | 0.38% | 3.19% | |
| MarginPolish | 0.33% | 2.25% | 0.26% | 2.84% | |
| HELEN | 2.09% | 17.30% | 0.48% | 19.88% | |
| NeuralPolish | 0.42% | 1.56% | 0.55% | 2.53% | |
| NA12878 chr20 | Flye | 0.33% | 3.08% | 0.27% | 3.68% |
| Racon | 0.31% | 2.99% | 0.36% | 3.66% | |
| Medaka | 0.35% | 2.50% | 0.32% | 3.16% | |
| MarginPolish | 0.28% | 2.32% | 0.32% | 2.92% | |
| HELEN | 2.11% | 16.83% | 0.59% | 19.53% | |
| NeuralPolish | 0.34% | 1.56% | 0.50% | 2.40% | |
| A.thaliana | Flye | 0.73% | 3.94% | 1.99% | 6.66% |
| Racon | 0.69% | 3.57% | 2.11% | 6.37% | |
| Medaka | 0.74% | 3.47% | 2.03% | 6.23% | |
| MarginPolish | 0.69% | 3.49% | 2.05% | 6.23% | |
| HELEN | 1.07% | 4.79% | 2.11% | 7.97% | |
| NeuralPolish | 0.73% | 2.94% | 2.37% | 6.04% |
| Dataset | Tool | Mismatch | Deletion | Insertion | Error rate |
|---|---|---|---|---|---|
| E.coli | Flye | 0.18% | 0.09% | 0.07% | 0.34% |
| Racon | 0.11% | 0.17% | 0.13% | 0.41% | |
| Medaka | 0.14% | 0.05% | 0.10% | 0.29% | |
| MarginPolish | 0.11% | 0.08% | 0.11% | 0.30% | |
| HELEN | 0.09% | 0.12% | 0.10% | 0.31% | |
| NeuralPolish | 0.07% | 0.10% | 0.08% | 0.25% | |
| S.cerevisiae | Flye | 0.10% | 0.75% | 0.50% | 1.35% |
| Racon | 0.12% | 0.75% | 0.63% | 1.50% | |
| Medaka | 0.10% | 0.54% | 0.52% | 1.16% | |
| MarginPolish | 0.09% | 0.60% | 0.40% | 1.09% | |
| HELEN | 0.09% | 0.74% | 0.44% | 1.26% | |
| NeuralPolish | 0.11% | 0.69% | 0.58% | 1.38% | |
| NA12878 chr21 | Flye | 0.39% | 2.90% | 0.32% | 3.61% |
| Racon | 0.37% | 2.84% | 0.42% | 3.63% | |
| Medaka | 0.42% | 2.38% | 0.38% | 3.19% | |
| MarginPolish | 0.33% | 2.25% | 0.26% | 2.84% | |
| HELEN | 2.09% | 17.30% | 0.48% | 19.88% | |
| NeuralPolish | 0.42% | 1.56% | 0.55% | 2.53% | |
| NA12878 chr20 | Flye | 0.33% | 3.08% | 0.27% | 3.68% |
| Racon | 0.31% | 2.99% | 0.36% | 3.66% | |
| Medaka | 0.35% | 2.50% | 0.32% | 3.16% | |
| MarginPolish | 0.28% | 2.32% | 0.32% | 2.92% | |
| HELEN | 2.11% | 16.83% | 0.59% | 19.53% | |
| NeuralPolish | 0.34% | 1.56% | 0.50% | 2.40% | |
| A.thaliana | Flye | 0.73% | 3.94% | 1.99% | 6.66% |
| Racon | 0.69% | 3.57% | 2.11% | 6.37% | |
| Medaka | 0.74% | 3.47% | 2.03% | 6.23% | |
| MarginPolish | 0.69% | 3.49% | 2.05% | 6.23% | |
| HELEN | 1.07% | 4.79% | 2.11% | 7.97% | |
| NeuralPolish | 0.73% | 2.94% | 2.37% | 6.04% |
For HELEN polishing, the polishing results of the NA12878 datasets are abnormal because the models of MarginPolish and HELEN do not support Albacore basecaller. So we downloaded the latest NA12878 raw reads (rel6, ONT Guppy basecaller v2.3.8) from the public website. We assembled the genomes of NA12878 chromosome 20 and 21 using Wtdbg2, Flye and Canu separately. Then we used MarginPolish, HELEN and NeuralPolish to polish the draft assemblies and evaluated the error rates of polished results by Pomoxis. The error rates of polished assembly are shown in Table 3. On five out of the six datasets, the error rates of polishing results by NeuralPolish are lower than that of polishing results by MarginPolish and HELEN. From the error types, in all datasets the errors are predominantly deletions, and NeuralPolish obtains the lowest deletion error rate in each dataset.
Comparison of the polishing error rates of MarginPolish, HELEN and NeuralPolish on the data of latest NA12878 rel6. The bold in the table means the best results.
| Dataset | Tool | Mismatch (%) | Deletion (%) | Insertion (%) | Error rate (%) |
|---|---|---|---|---|---|
| NA12878 chr21 | MarginPolish | 0.188 | 0.527 | 0.341 | 1.056 |
| (Wtdbg2) | HELEN | 0.194 | 0.586 | 0.347 | 1.127 |
| NeuralPolish | 0.217 | 0.444 | 0.347 | 1.008 | |
| NA12878 chr20 | MarginPolish | 0.134 | 0.574 | 0.316 | 1.024 |
| (Wtdbg2) | HELEN | 0.129 | 0.684 | 0.3 | 1.113 |
| NeuralPolish | 0.162 | 0.536 | 0.318 | 1.016 | |
| NA12878 chr21 | MarginPolish | 0.257 | 0.486 | 0.43 | 1.173 |
| (Flye) | HELEN | 0.246 | 0.509 | 0.373 | 1.128 |
| NeuralPolish | 0.284 | 0.422 | 0.412 | 1.118 | |
| NA12878 chr20 | MarginPolish | 0.193 | 0.504 | 0.371 | 1.068 |
| (Flye) | HELEN | 0.177 | 0.508 | 0.34 | 1.025 |
| NeuralPolish | 0.225 | 0.449 | 0.405 | 1.079 | |
| NA12878 chr21 | MarginPolish | 0.258 | 0.632 | 0.379 | 1.269 |
| (Canu) | HELEN | 0.278 | 0.702 | 0.353 | 1.333 |
| NeuralPolish | 0.278 | 0.611 | 0.371 | 1.260 | |
| NA12878 chr20 | MarginPolish | 0.157 | 0.522 | 0.335 | 1.014 |
| (Canu) | HELEN | 0.153 | 0.564 | 0.318 | 1.035 |
| NeuralPolish | 0.185 | 0.454 | 0.356 | 0.995 |
| Dataset | Tool | Mismatch (%) | Deletion (%) | Insertion (%) | Error rate (%) |
|---|---|---|---|---|---|
| NA12878 chr21 | MarginPolish | 0.188 | 0.527 | 0.341 | 1.056 |
| (Wtdbg2) | HELEN | 0.194 | 0.586 | 0.347 | 1.127 |
| NeuralPolish | 0.217 | 0.444 | 0.347 | 1.008 | |
| NA12878 chr20 | MarginPolish | 0.134 | 0.574 | 0.316 | 1.024 |
| (Wtdbg2) | HELEN | 0.129 | 0.684 | 0.3 | 1.113 |
| NeuralPolish | 0.162 | 0.536 | 0.318 | 1.016 | |
| NA12878 chr21 | MarginPolish | 0.257 | 0.486 | 0.43 | 1.173 |
| (Flye) | HELEN | 0.246 | 0.509 | 0.373 | 1.128 |
| NeuralPolish | 0.284 | 0.422 | 0.412 | 1.118 | |
| NA12878 chr20 | MarginPolish | 0.193 | 0.504 | 0.371 | 1.068 |
| (Flye) | HELEN | 0.177 | 0.508 | 0.34 | 1.025 |
| NeuralPolish | 0.225 | 0.449 | 0.405 | 1.079 | |
| NA12878 chr21 | MarginPolish | 0.258 | 0.632 | 0.379 | 1.269 |
| (Canu) | HELEN | 0.278 | 0.702 | 0.353 | 1.333 |
| NeuralPolish | 0.278 | 0.611 | 0.371 | 1.260 | |
| NA12878 chr20 | MarginPolish | 0.157 | 0.522 | 0.335 | 1.014 |
| (Canu) | HELEN | 0.153 | 0.564 | 0.318 | 1.035 |
| NeuralPolish | 0.185 | 0.454 | 0.356 | 0.995 |
Note: The draft assemblies of NA12878 chromosome 20 and 21 were assembled by Wtdbg2, Flye and Canu.
Comparison of the polishing error rates of MarginPolish, HELEN and NeuralPolish on the data of latest NA12878 rel6. The bold in the table means the best results.
| Dataset | Tool | Mismatch (%) | Deletion (%) | Insertion (%) | Error rate (%) |
|---|---|---|---|---|---|
| NA12878 chr21 | MarginPolish | 0.188 | 0.527 | 0.341 | 1.056 |
| (Wtdbg2) | HELEN | 0.194 | 0.586 | 0.347 | 1.127 |
| NeuralPolish | 0.217 | 0.444 | 0.347 | 1.008 | |
| NA12878 chr20 | MarginPolish | 0.134 | 0.574 | 0.316 | 1.024 |
| (Wtdbg2) | HELEN | 0.129 | 0.684 | 0.3 | 1.113 |
| NeuralPolish | 0.162 | 0.536 | 0.318 | 1.016 | |
| NA12878 chr21 | MarginPolish | 0.257 | 0.486 | 0.43 | 1.173 |
| (Flye) | HELEN | 0.246 | 0.509 | 0.373 | 1.128 |
| NeuralPolish | 0.284 | 0.422 | 0.412 | 1.118 | |
| NA12878 chr20 | MarginPolish | 0.193 | 0.504 | 0.371 | 1.068 |
| (Flye) | HELEN | 0.177 | 0.508 | 0.34 | 1.025 |
| NeuralPolish | 0.225 | 0.449 | 0.405 | 1.079 | |
| NA12878 chr21 | MarginPolish | 0.258 | 0.632 | 0.379 | 1.269 |
| (Canu) | HELEN | 0.278 | 0.702 | 0.353 | 1.333 |
| NeuralPolish | 0.278 | 0.611 | 0.371 | 1.260 | |
| NA12878 chr20 | MarginPolish | 0.157 | 0.522 | 0.335 | 1.014 |
| (Canu) | HELEN | 0.153 | 0.564 | 0.318 | 1.035 |
| NeuralPolish | 0.185 | 0.454 | 0.356 | 0.995 |
| Dataset | Tool | Mismatch (%) | Deletion (%) | Insertion (%) | Error rate (%) |
|---|---|---|---|---|---|
| NA12878 chr21 | MarginPolish | 0.188 | 0.527 | 0.341 | 1.056 |
| (Wtdbg2) | HELEN | 0.194 | 0.586 | 0.347 | 1.127 |
| NeuralPolish | 0.217 | 0.444 | 0.347 | 1.008 | |
| NA12878 chr20 | MarginPolish | 0.134 | 0.574 | 0.316 | 1.024 |
| (Wtdbg2) | HELEN | 0.129 | 0.684 | 0.3 | 1.113 |
| NeuralPolish | 0.162 | 0.536 | 0.318 | 1.016 | |
| NA12878 chr21 | MarginPolish | 0.257 | 0.486 | 0.43 | 1.173 |
| (Flye) | HELEN | 0.246 | 0.509 | 0.373 | 1.128 |
| NeuralPolish | 0.284 | 0.422 | 0.412 | 1.118 | |
| NA12878 chr20 | MarginPolish | 0.193 | 0.504 | 0.371 | 1.068 |
| (Flye) | HELEN | 0.177 | 0.508 | 0.34 | 1.025 |
| NeuralPolish | 0.225 | 0.449 | 0.405 | 1.079 | |
| NA12878 chr21 | MarginPolish | 0.258 | 0.632 | 0.379 | 1.269 |
| (Canu) | HELEN | 0.278 | 0.702 | 0.353 | 1.333 |
| NeuralPolish | 0.278 | 0.611 | 0.371 | 1.260 | |
| NA12878 chr20 | MarginPolish | 0.157 | 0.522 | 0.335 | 1.014 |
| (Canu) | HELEN | 0.153 | 0.564 | 0.318 | 1.035 |
| NeuralPolish | 0.185 | 0.454 | 0.356 | 0.995 |
Note: The draft assemblies of NA12878 chromosome 20 and 21 were assembled by Wtdbg2, Flye and Canu.
3.3 Quast evaluation for polished assembly
Quast is a widely used quality assessment tool for genome assemblies. We used Quast to evaluate the quality of the assemblies polished by different polishing tools. The draft assemblies are assembled with Flye. To calculate the assembly identity, we divided each contig into the size of 10 kb. Each divided piece is aligned to the reference genome by minimap2. The identity is defined as the number of matching bases divided by the alignment length. The final assembly identity is the median of the identities of these 10 kb assembly segments. The Quast reports and assembly identities of polished results are shown in Supplementary Table S3 (Quast reports and assembly identities of polishing Wtdbg2 assemblies and Canu assemblies are provided in Supplementary Tables S4 and S5). The N50 of polished contigs by NeuralPolish is similar to that of polished contigs by other polishing tools. On the data of NA12878 chromosome 21, chromosome 20 and A.thaliana, NeuralPolish gets the least indel errors per 100k base pairs. Meanwhile, the number of indels longer than 5 bp in polished contigs by NeuralPolish is also smaller than that by other polishing tools. For Medaka, it is able to reduce the number of indels per 100k base pairs, but there is no significant reduction in the number of indels longer than 5 bp. It means that Medaka can only solve short indel errors (≤5 bp), but NeuralPolish can deal with both short and longer ones (>5 bp). In terms of genome identity, NeuralPolish has the highest identity on four out of the five test datasets. On the test data of NA12878 chromosomes 21 and chromosome 20, the increase in genome identity by NeuralPolish is the most significant.
3.4 Analysis of error sites in the assembly polished by NeuralPolish
To better understand the difference between contigs before and after NeuralPolish polishing, we compared the draft assembly with the polished assembly in terms of error sites. The draft assembly was assembled from raw reads of NA12878 chromosome 20 by Flye. Then we used Mummer (Marçais et al., 2018) to align the draft assembly and polished assembly to the reference to find the error sites. Analysis of the error sites in draft assembly and polished assembly is shown in Supplementary Figure S2. In terms of deletion errors, a total of 1 171 405 deletion sites in the draft assembly were detected and corrected, and 614 127 deletion sites remained in the polished assembly. During the correction process of NeuralPolish, some new deletion errors were introduced. The number of newly introduced error sites was 256 174, accounting for 29.44% of the total deletion errors. In the polished assembly generated by NeuralPolish, the number of deleted error sites was reduced from 1 785 532 to 870 301. For insertion errors, the draft assembly contains 12 825 insertion error sites. NeuralPolish identified and corrected 8 387 error sites, accounting for 65%. But NeuralPolish introduced 101 982 insertion error sites. Compared with the draft assembly by Flye, the number of insertion error sites in the polished assembly was increased from 12 825 to 106 420. The reason for the increase of insertion errors is when NeuralPolish corrected a large number of deletion errors, some correct sites were incorrectly detected as deletion error sites and then NeuralPolish modified these correct sites. For substitution errors, there were 153 668 error sites in draft assembly. NeuralPolish identified and corrected 82 877 error sites, accounting for 54%. During the correction process of NeuralPolish, 75 649 new substitution errors were introduced. Compared with the draft assembly, the number of substitution error sites in the assembly polished by NeuralPolish was reduced from 153 668 to 146 440. In general, in the NeuralPolish polishing of NA12878 chromosome 20, NeuralPolish solved a large number of deletion errors at the cost of introducing some new insertion errors. Since the number of solved sites is more than three times the number of newly introduced error sites, the total number of error sites in the polished assembly still decreased significantly.
3.5 Counting homopolymer 5-mers in polished assembly and reference genome
Due to the mechanism of Nanopore sequencing, indel errors are more likely to occur in the homopolymers. We chose the homopolymer 5-mers consisting of a single (e.g. AAAAA) base or 4-base stretch of a single base (e.g. AAAAT). Then we compared the number of these 5-mers appeared in the contigs with that in the reference. We selected the homopolymer 5-mers with the top 20 occurrence frequency in the reference, and the numbers of these 5-mers in the polished assembly are presented in Supplementary Figure S3. Comparing NeuralPolish with Racon, Medaka and MarginPolish, the number of homopolymer 5-mers in NeuralPolish polished assembly is much closer to the real value (the number of homopolymer 5-mers in the reference) in almost every chosen homopolymer 5-mer. Besides, there are seven types of homopolymer 5-mers, the numbers of which in the HELEN polished assembly are far from the real value, whereas the numbers in NeuralPolish polished assembly are consistently close to the real values across all homopolymer 5-mers.
3.6 Evaluation of NeuralPolish on assemblies with varying degree of errors
Currently, a variety of basecallers are available for converting Nanopore raw signals to DNA sequences. The most commonly used basecallers are Albacore and Guppy which are developed by ONT. These basecallers are constantly updated and different versions have varying raw read identities. As a result, the error rates of draft assemblies are also different. To evaluate NeuralPolish on the assemblies with different error rates, we used four read sets of NA12878 chromosome 21 which were basecalled by Albacore v2.3.4, Guppy v2.3.8, Guppy v4.4.0 fast model and Guppy v4.4.0 high accuracy (hac) model separately. Then we used Flye to assemble these read sets and polished the draft assemblies using NeuralPolish. The read identity of each read set and the error rates of the polishing results are shown in Supplementary Figure S4. The median identities of read sets range from 83.4% to 92.52%. And the error rates of polished assemblies range from 1.29% to 3.61%. In all datasets with varying degree of errors, NeuralPolish reduced the error rate of the assemblies. When the reads were basecalled by latest basecaller Guppy v4.4.0 high accuracy model, the polished assembly produced by NeuralPolish had a relative improvement rate of 23.5% over the draft assembly in terms of the error rate. Overall, NeuralPolish can be used to polishing the assemblies of varying degree of errors as well as the datasets generated by different basecallers.
3.7 Polishing assemblies with multiple rounds of NeuralPolish
When correcting the assembly using polishers, multiple rounds of polishing often lead to a more accurate assembly. To evaluate the performance of NeuralPolish iterative polishing, we performed four rounds of NeuralPolish on the five test datasets. In the iterative polishing, we only ran Racon during the first round of NeuralPolish. In subsequent iterations, we aligned the raw reads to the polishing result from previous iteration and then predicted the polished contigs of current iteration. The error rates of the iterative polishing results are provided in Supplementary Table S6. In four out of the five test datasets, NeuralPolish iterative polishing further reduces the errors in the assemblies. In the dataset of NA12878 chromosome 21, three iterations of NeuralPolish lead to a error reduction of 0.07%. In the dataset of NA12878 chromosome 20, four iterations of NeuralPolish have a further error reduction of 0.02%. In the dataset of A.thaliana, four iterations of NeuralPolish reduce the error rate of the assembly from 6.06% to 5.95%. Overall, when polishing assemblies using NeuralPolish, iterative polishing can achieve more accurate assemblies.
3.8 Computational setup and runtime
There are two main steps in NeuralPolish, alignment matrix construction and neural network prediction. The alignment matrix construction is done on the server with 40 CPU processors [Intel(R) Xeon(R) Gold 6230 CPU @ 2.10 GHz]. The polished sequence prediction is performed on a Nvidia GeForce RTX 2080Ti GPU. Medaka and MarginPolish & HELEN are similar to NeuralPolish where part of the computation is on CPU and the rest is on GPU. Therefore, the overall runtime is obtained by adding the GPU runtime and the CPU runtime. The runtime of each polishing method performed on the data assembled with Flye is provided in Supplementary Table S7. In Racon polishing, we performed four iterations of Racon. In Medaka polishing, we performed four iterations of Racon followed by Medaka. In MarginPolish and HELEN polishing, we ran MarginPolish first and then HELEN. In NeuralPolish polishing, we performed one round of Racon follow by NeuralPolish. When the coverage of raw reads exceeds 40×, NeuralPolish will randomly pick at most 40× of them, while other polishing methods will use all the data. So in the data of E.coli (319×) and S.cerevisiae (512×), NeuralPolish takes the least time. When the species is relatively complex, the running time of NeuralPolish will be longer than that of other tools. However, the extra time spent enables NeuralPolish to obtain more accurate polishing results. NeuralPolish takes more runtime on the datasets of NA12878 chromosome 21, NA12878 chromosome 20 and A.thaliana, but it also achieves the lowest error rates on these datasets. The reason why NeuralPolish takes more time when polishing complex species is that there is a CTC algorithm used in NeuralPolish. For a given input feature, the CTC decoder calculates all possible output distributions. When the species is more complex, there are a much larger number of types of output distributions.
4 Conclusion
In this study, we proposed a novel Nanopore polishing method, NeuralPolish, which generates polished assemblies with fewer errors on a wide range of test datasets compared to Racon, Medaka, MarginPolish and HELEN. In NeuralPolish, we designed an alignment matrix construction from read-to-assembly alignment. In the network architecture, NeuralPolish uses two orthogonal bi-directional GRU networks to process the matrix by row and column, respectively. Then the final polished sequence is processed by a CTC decoder with a greedy algorithm. Our NeuralPolish model is trained on the assembly produced by Wtdbg2, and it outperforms Racon, Medaka, MarginPolish and HELEN in terms of the assembly error rate on most of the test datasets. Besides, we evaluated NeuralPolish on the assembly generated by Flye and Canu, NeuralPolish obtains the polished assemblies with fewer errors. From the analysis of error sites of the polished assembly, we found that NeuralPolish solves a large number of deletion errors at the cost of introducing some insertion errors, thereby reducing the overall error rate of the draft assembly. From the comparison of the homopolymer 5-mers counts in the polished genome assembly and reference genome on the data of NA12878 chromosome 20, we found that the distribution of 5-mers in the assembly polished by NeuralPolish is closer to that of the reference in the homopolymer regions.
NeuralPolish is a haploid polishing method, and thus it can not recognize and solve heterozygous polymorphisms. Since the error distribution and the error rate of PacBio sequencing data is different from that of Nanopore sequencing data, we will further develop NeuralPolish to support the polishing of PacBio assembly in future work.
Funding
This work was supported in part by the National Natural Science Foundation of China under Grants [U1909208 and 61772557], 111 Project [B18059], Hunan Provincial Science and Technology Program [2018wk4001 to J.W.], the U. S. National Institute of Food and Agriculture (NIFA) [2017-70016-26051] and the U.S.National Science Foundation (NSF) [ABI-1759856 to F.L.], and the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. [FCC/1/1976-26-01, URF/1/3412-01-01, URF/1/4098-01-01 and REI/1/4473-01-01 to X.G.].
Conflict of Interest: none declared.


{kind=link}
{kind=link}