





Abstract
‘PoseFilter’ is a PyMOL plugin that assists in analyses and filtering of docked poses. PoseFilter enables automatic detection of symmetric poses from docking outputs and can be accessed using both graphical user interface and command-line options within the PyMOL program. Two methods of analyses, root mean square deviations and interaction fingerprints, are available from this plugin. The capabilities of the plugin are demonstrated using docking outputs from different oligomeric protein-ligand complexes.
The plugin can be downloaded from the GitHub page, https://github.com/skalyaanamoorthy/PoseFilter.
Supplementary data are available at Bioinformatics online.
1 Introduction
Oligomerization is a common characteristic in many biomolecules such as proteins, especially, enzymes, ion channels and G-protein coupled receptors (Forrest, 2015; Wilding et al., 2017). Oligomers create symmetry in the molecule, which could result in a binding site made of the identical residues from adjacent monomeric units. Molecular docking is a commonly used computational technique for predicting the binding mode and interactions between molecules. Docking on symmetrical binding sites often results in multiple symmetrical binding poses that exhibit similar binding modes but on the adjacent monomers (Kalyaanamoorthy and Barakat, 2018). Manual post-processing of docked poses for removing redundant poses and identifying similar binding modes (Schiesaro et al., 2013) through interactions is time-consuming, especially when performing screening of large ligand libraries to an oligomeric target (Schiesaro et al., 2013). Therefore, we have developed a new application called ‘PoseFilter’ as a plugin to the widely used PyMOL package (Schrödinger, 2021) to perform post-processing of docked complexes.
2 Implementation
‘PoseFilter’ is implemented using Python language as a plugin in the PyMOL program (Schrödinger, 2021) and can be accessed using both a graphical user and command line interface . PoseFilter reads a directory containing protein and multiple docked poses in any PyMOL-compatible format of the ligand and self-determines the type of n-mer (dimer, trimer, etc.). Users can analyze their docked results in two ways: (i) root mean square deviation (RMSD)-based analysis; (ii) interaction fingerprint-based analysis and identify which of the poses are similar and unique among the docked solutions.
2.1 RMSD analysis
PoseFilter’s RMSD processing differs from other RMSD scoring programs by providing functionality to process each submitted pose as reference structure that allows all to all comparison of the binding conformations and also differentiate any symmetrical poses when the ligands bind to oligomers. Symmetrical poses are identified through a normalization process where monomers are rotated, positioning ligands onto the same subunit as the reference. PoseFilter performs one-to-one comparisons of each docked pose with every other pose.
For example, a trimer (n = 3, Fig. 1A) with chains A, B, C will be rotated three times and fitted to the reference structure using PyMOL’s ‘pair_fit’ command. As shown in Figure 1B, the reference (bolded) and comparison structures (unbolded) both have chains A, B, C. The reference remains the same throughout each operation. Each ‘pair_fit’ operation in this trimer example rotates the structure clockwise by 0, 1 and 2 times and the corresponding ligand RMSD is calculated using cmd.rms_cur(ligand, reference). The rotation that gives the smallest RMSD between the ligand pose and the reference pose is selected. The pose files are then sorted based on the RMSD of the rotated ligand poses. A default threshold of 2.0 Å is set to classify poses as ‘Similar’ (≤2.0 Å) and ‘Unique’ (>2.0 Å), which can be modified by the user.
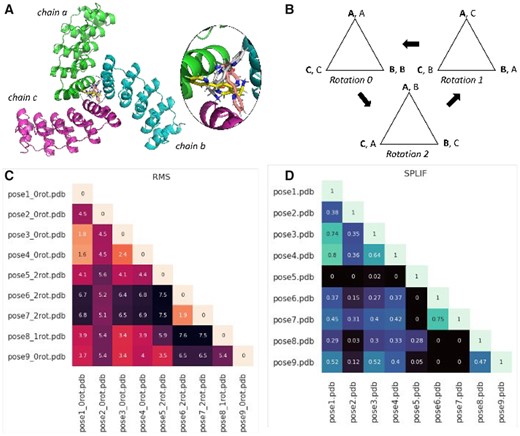
Snapshot of the results from PoseFilter using a trimer as an example. (A) Cartoon representation of trimeric metalloprotein protein bound to 3-(2,2′-bipyridin-5-Yl)-l-Alanine (PDB id: 5EIL). (B) Diagrammatic representation of the rotations performed in homo-trimeric states. (C) Heatmap showing the minimum RMSD of the poses among the different rotations (lower RMSD values indicate more similar poses). (D) Heatmap showing the similarity between the poses as assessed by SPLIF fingerprint (1 represents identical pose and 0 represents no similarity)
2.2 Fingerprint analysis
Interaction fingerprint-based sorting identifies poses that show a similar interaction pattern with amino acid residues in the binding site. Two fingerprint options were implemented, interaction fingerprint and SPLIF fingerprint, as offered in the ‘Open Drug Discovery Toolkit’ (oddt) package (Wójcikowski et al., 2015). The simple interaction fingerprint encodes bytes (0-8) for each protein residue, based on presence or absence of an interaction, which are then grouped to create a fixed-size fingerprint (Chupakhin et al., 2014). Alternatively, the SPLIF fingerprints find matching circular segments, return a list of 2D ‘matches’ and then extracts their 3D coordinates to estimate their RMSD and check if there is a match based on a fixed criterion (Da and Kireev, 2014). For each of these fingerprints, oddt provides functions to give a 0-1 score for comparison of two ligands. A default threshold of 0.5 is set to classify the poses as ‘Unique’ (≤0.5) and ‘Similar’ (>0.5), which can be modified to user’s preference.
3 Results
The RMSD and fingerprint analysis are performed by comparing each pose to every other pose. A matrix of RMSD and fingerprint values is written to a comma-separated (.CSV) file, and a lower triangle heatmap is created to enable comprehensive visual analysis of the results from all docked poses. A residue interaction report is written out using the oddt interaction module that characterizes eight different interactions (hydrophobic, face to face and edge to face aromatic contacts, hydrogen bond interactions and salt bridge interactions) formed between the ligand and the protein.
4 Case study
We used a homo-trimeric metalloprotein (PDB id: 5EIL) to demonstrate the PoseFilter functionality (Fig. 1A). The ligand found in the crystal complex was redocked to its target using MOE program using default settings (Wójcikowski et al., 2015). Docked poses were used as input for the RMSD and fingerprint analysis. A 2.0 Å threshold was used for the RMSD analysis to classify similar and dissimilar poses. The ‘_nrot’ suffix added to labels shows which rotation gave the lowest RMS value. Poses showing lighter shades in the heatmap, poses 6 (rotation 2) and 7 (rotation 2); poses 1, 3 and 4 (rotation 1) are similar with RMSD values of 1.9 Å, 1.7 Å and 1.6 Å, respectively (Fig. 1C). Similarly, the fingerprint analysis carried out with a cutoff value of 0.5, showed the poses listed above (poses 6,7 and poses 1, 3, 4) to have a higher fingerprint score of >0.7 (Fig. 1D), illustrating similar modes of binding. The two types of analyses allow the users to analyze complexes from both rigid and flexible docking. Examples of dimer and tetramer analyses are provided in Supporting Information.
5 Conclusion
PoseFilter program provides a user-friendly graphical/command-line interface to compare, filter and analyze docking results. This plugin is useful for detecting redundant and symmetrical poses from docking results. The RMS and interaction fingerprint analyses provide different ways to compare and analyze complexes from rigid and flexible/ensemble docking and therefore, can be useful for post-processing docking outputs.
Acknowledgement
The authors thank Dr Maciek Wójcikowski for discussion on SPLIF fingerprints.
Funding
Authors thank the University of Waterloo’s Undergraduate Research Internship (URI) support to Justine C. Williams. This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund (Grant #CFREF-2015-00011).
Conflict of Interest: none declared.


{kind=link}