





Abstract
Species tree estimation from genes sampled from throughout the whole genome is complicated due to the gene tree–species tree discordance. Incomplete lineage sorting (ILS) is one of the most frequent causes for this discordance, where alleles can coexist in populations for periods that may span several speciation events. Quartet-based summary methods for estimating species trees from a collection of gene trees are becoming popular due to their high accuracy and statistical guarantee under ILS. Generating quartets with appropriate weights, where weights correspond to the relative importance of quartets, and subsequently amalgamating the weighted quartets to infer a single coherent species tree can allow for a statistically consistent way of estimating species trees. However, handling weighted quartets is challenging.
We propose wQFM, a highly accurate method for species tree estimation from multi-locus data, by extending the quartet FM (QFM) algorithm to a weighted setting. wQFM was assessed on a collection of simulated and real biological datasets, including the avian phylogenomic dataset, which is one of the largest phylogenomic datasets to date. We compared wQFM with wQMC, which is the best alternate method for weighted quartet amalgamation, and with ASTRAL, which is one of the most accurate and widely used coalescent-based species tree estimation methods. Our results suggest that wQFM matches or improves upon the accuracy of wQMC and ASTRAL.
Datasets studied in this article and wQFM (in open-source form) are available at https://github.com/Mahim1997/wQFM-2020.
Supplementary data are available at Bioinformatics online.
1 Introduction
With the rapid growth rate of newly-sequenced genomes, it is now common to estimate species trees from genes sampled throughout the whole genome. However, each individual gene has its own phylogeny (known as a gene tree), which may not agree with the species tree. Species tree estimation from multi-locus datasets is complicated in the presence of species tree–gene tree heterogeneity, which can result from many biological processes, of which incomplete lineage sorting (ILS), modeled by the multi-species coalescent (MSC) (Kingman, 1982), is probably the most common. ILS is also known as ‘deep coalescence’, which occurs with high probability whenever the time between speciation events is short relative to the population size (Maddison, 1997). ILS presents substantial challenges to species tree estimation (Degnan and Rosenberg, 2009; Edwards, 2009). For example, the standard approach, concatenation (which concatenates multiple sequence alignments of different genes into a single super-alignment and then estimates a tree from this alignment) can be statistically inconsistent (Roch and Steel, 2015) and can return incorrect trees with high confidence (DeGiorgio and Degnan, 2010; Edwards et al., 2007; Kubatko and Degnan, 2007; Leaché and Rannala, 2011). Moreover, under some conditions, the most probable gene tree topology may not agree with the species tree, which is known as the ‘anomaly zone’ (Degnan, 2013; Degnan and Rosenberg, 2009).
As a result of these studies, ‘summary methods’, which operate by computing gene trees from different loci and then combine the inferred gene trees into a species tree, are becoming increasingly popular (Bayzid and Warnow, 2013), and many of these summary approaches are statistically consistent under the MSC model (Bryant et al., 2012; Chifman and Kubatko, 2014; Heled and Drummond, 2010; Islam et al., 2020; Kubatko et al., 2009; Larget et al., 2010; Liu et al., 2009, 2010; Liu and Yu, 2011; Mirarab et al., 2014a; Mossel and Roch, 2010; Vachaspati and Warnow, 2015). Using the most basic pieces of phylogenetic information (i.e. triplets in a rooted setting, and quartets in an unrooted setting) is key to the design of some of the statistically consistent methods (Chifman and Kubatko, 2014; Mirarab et al., 2014a; Reaz et al., 2014). ASTRAL, which is one of the most accurate and widely used coalescent-based methods, tries to infer a species tree so that the number of induced quartets in the gene trees that are consistent with the species tree is maximized. Another approach is to infer individual quartets, and then amalgamate these quartets into a single coherent species tree (Avni et al., 2015; Chifman and Kubatko, 2014; Ma et al., 2008; Ranwez and Gascuel, 2001; Reaz et al., 2014; Schmidt et al., 2002; Snir and Rao, 2010; Strimmer and von Haeseler, 1996).
Quartet amalgamation techniques have drawn substantial interest both from practical and theoretical perspectives as quartets can be inferred from raw data with high accuracy and these quartets can subsequently be amalgamated to obtain a highly accurate species tree (Bryant and Steel, 2001; Chifman and Kubatko, 2014; Holland et al., 2013; Strimmer and von Haeseler, 1996). The summary methods are usually sensitive to gene tree estimation error (Bayzid and Warnow, 2013; Gatesy and Springer, 2014; Huang et al., 2010), so methods that can estimate species trees without needing to compute gene trees are of utmost importance. Therefore, quartet amalgamation techniques have attracted a lot of interest among the systematists. For example, SVDquartets (Chifman and Kubatko, 2014), using techniques from algebraic statistics, generates a set of quartets likely to be present in the species tree directly from sequence data; those quartets must then be assembled to infer a species tree. However, a given set of quartets may not exhibit perfect agreement and must therefore be assembled using a quartet assembly method like quartet Fiduccia–Mattheyses (QFM). Moreover, estimating a tree by finding the largest compatible subset of a given quartet set is computationally hard (Steel, 1992). Furthermore, this approach implicitly gives the same importance to all quartets, and thus does not take the relative reliabilities of various quartets into account (Ranwez and Gascuel, 2001).
There is ample evidence that assigning weights to quartets (where weight of a quartet denotes the relative confidence of a particular quartet topology out of the three alternate topologies on a set of four taxa) can improve phylogenetic analyses (Berry and Gascuel, 2000; Holland et al., 2013; Ranwez and Gascuel, 2001). This growing awareness about the importance of weighted quartets has led to the development of methods like wQMC (Avni et al., 2015), which is a weighted extension of the quartet max-cut (QMC) algorithm (Snir and Rao, 2010). In this article, we present a new method for amalgamating weighted quartets by extending QFM algorithm (Reaz et al., 2014), which is being widely used in important phylogenetic studies (Devitt et al., 2019; Hodel et al., 2018; Hosner et al., 2016; Kato et al., 2020; Malinsky et al., 2018; Mason et al., 2016; Moumi et al., 2019; Vázquez-Miranda et al., 2017; Yoder et al., 2016), especially along with SVDquartets (Chifman and Kubatko, 2014, 2015). SVDquartets has been implemented in PAUP* (Swofford, 2002), which uses QFM as a quartet agglomeration technique (Chou et al., 2015). We report, on an extensive evaluation study using widely used simulated dataset as well as biological dataset, the performance of wQFM. We have re-analyzed a collection of biological dataset, including the avian phylogenomic dataset (Jarvis et al., 2014) comprising 14 446 genes across 48 genomes representing all avian orders. Our results suggest that wQFM achieves competitive or—in many cases—better tree accuracy than the main two alternatives, wQMC and ASTRAL. Notably, the avian tree estimated by wQFM is more consistent with established hypotheses than the tree estimated by ASTRAL.
2 Approach
We consider the weighted version of the Maximum Quartet Consistency (MQC) problem (Reaz et al., 2014; Snir and Rao, 2010), which we call , and define as follows.
| Problem | Weighted Maximum Quartet Consistency () |
| Input | A set of weighted quartets on a set of taxa. |
| Output | A tree T on such that the total weight of the quartets in that are consistent with T is maximized. |
| Problem | Weighted Maximum Quartet Consistency () |
| Input | A set of weighted quartets on a set of taxa. |
| Output | A tree T on such that the total weight of the quartets in that are consistent with T is maximized. |
| Problem | Weighted Maximum Quartet Consistency () |
| Input | A set of weighted quartets on a set of taxa. |
| Output | A tree T on such that the total weight of the quartets in that are consistent with T is maximized. |
| Problem | Weighted Maximum Quartet Consistency () |
| Input | A set of weighted quartets on a set of taxa. |
| Output | A tree T on such that the total weight of the quartets in that are consistent with T is maximized. |
wQFM is an extension of the QFM algorithm to solve the problem. In this study, we especially considered the problem in the context of species tree estimation from a collection of gene trees, where the weight of a quartet q is computed based on the number of that induce quartet topology q. In this context, we call this method wQFM-GTF [i.e. wQFM with quartets weighted based on gene tree frequencies (GTF)]. wQFM-GTF uses a two-step technique in which we first use the input set of estimated gene trees to produce a set of weighted four-taxon trees (called ‘weighted quartet trees’, or ‘weighted quartets’), and then combine these weighted quartet trees into a tree on the full set of taxa using a heuristic aimed at finding a species tree of minimum distance to the set of weighted quartet trees (details below). Thus, wQFM-GTF is similar in overall structure to the population tree in BUCKy (Ané et al., 2007; Larget et al., 2010). To understand wQFM, we begin by defining a very simple approach, Combining Dominant Quartet Trees (), which is a statistically consistent approach. The proof that is statistically consistent provides intuition as to why the solution to problem is statistically consistent, and motivates its algorithmic design.
2.1 Combining Dominant Quartet Trees
The basic idea is to take the input set of gene trees, compute a ‘dominant quartet tree’ (i.e. the most frequent quartet topology) for every four species, and then combine the dominant quartet trees into a supertree on the full set of species using a preferred quartet amalgamation technique. If the quartet amalgamation technique correctly computes the supertree when the dominant quartet trees are ‘compatible’ (see below), then is a statistically consistent method under the MSC. Thus, depends on the quartet amalgamation technique, and so is a general technique, and not a particular technique.
Because the method depends on the choice of quartet amalgamation technique, we now discuss this issue. We say that a set of quartet trees is ‘compatible’ if there is a tree T such that every quartet tree is topologically identical to the subtree of T induced on its leaf set. Furthermore, when the set of quartet trees is compatible, then there is a unique tree that induces all the quartet trees (called the ‘compatibility supertree’), and it can be computed in polynomial time using very simple techniques [e.g. the Naive Quartet Method, discussed in Erdos et al. (1999)]. Thus, while there are many quartet amalgamation techniques, most of them are able to return the compatibility supertree when the input set contains a tree on every four leaves and is compatible. We call such quartet amalgamation techniques ‘proper’.
If the quartet amalgamation technique is proper, then is statistically consistent under the MSC model.
See Theorem 2 in Davidson et al. (2015).
If the quartet amalgamation technique is proper and the weights of the input quartets are computed based on GTF, then the unique solution to the problem is a statistically consistent estimator of the true species tree under the MSC model.
This was proved as a part of Theorem 2 in Mirarab et al. (2014a), but we provide an outline of the proof here for the sake of completeness. Suppose, we are given a large number of true gene trees so that the most probable gene tree is also the dominant gene tree. Therefore, the weights on quartet trees will be equal to the proportion of the gene trees that induce that particular quartet tree. Note that the weight of the dominant quartet tree will be greater than all other quartet trees. Because that the dominant quartet tree (one with the highest frequency) is the most probable gene tree, and hence also the true species tree for its leaf set (since there are no anomalous 4-leaf unrooted gene trees), the best score is obtained by the true species tree.
The proof that and are statistically consistent under the MSC model provides a guarantee under idealized conditions—where all gene trees are correct and there are a sufficiently large number of them. However, in practice, estimated gene trees have error and there may not be a sufficiently large number of gene trees. Therefore, for good performance (and not just theoretical guarantees), species tree estimation methods need to work well with estimated gene trees—for which the dominant gene trees may not be identical to the most probable gene trees, and hence may not be compatible with each other. Therefore, heuristics for combining quartet trees, that can construct supertrees even when the quartet trees are incompatible, are valuable techniques for species tree estimation in the presence of ILS.
2.2 Related methods
BUCKy-pop (Larget et al., 2010) (the population tree output by BUCKy) is one of the statistically consistent methods for species tree estimation under the MSC model that uses a quartet-based approach. The input to BUCKy is a set of unrooted gene tree distributions, with one distribution per gene [BUCKy was originally intended for use with posterior distributions computed using Bayesian MCMC methods, but has also been used with distributions computed using maximum likelihood (ML) bootstrapping; both approaches give similar results (Yang and Warnow, 2011)]. In the first step, BUCKy-pop uses the gene tree distributions to estimate a quartet tree for every four species, and performs this estimation using Bayesian techniques. In the second step, it combines these estimated quartet trees using a quartet tree amalgamation technique (Ma et al., 2008). Because the quartet tree amalgamation technique will reconstruct the compatibility supertree if it exists, BUCKy-pop is statistically consistent under the MSC model. Another popular method is SVDquartets (Chifman and Kubatko, 2014, 2015), where the loci in a multi-locus dataset are concatenated into a single long alignment, and then, for each set of four species, a quartet tree for that set is computed using algebraic statistics and singular value decomposition. Finally, a species tree is estimated (using QFM or QMC) by amalgamating the quartet trees so that it agrees with as many of these quartet trees as possible. ASTRAL, another quartet-based method, uses dynamic programming to find an optimal tree within a constrained version of tree space defined by the input gene trees. Its optimization problem is based on the weighted quartet score of a candidate species tree defined to be the number of quartets from the set of input gene trees that agree with the candidate species tree.
2.3 wQFM and wQFM-GTF
We present a new summary method, wQFM-GTF, which estimates species trees by combining the quartets induced by the gene trees. However, unlike SVDquartets, BUCKy-pop and , which consider a single quartet for each set of four taxa and compute species trees by combining these ‘dominant’ quartet trees, wQFM-GTF computes weights for every possible four-leaf tree (and so for each of the three possible unrooted trees for every four leaves), and then combines this set of weighted quartet trees into a tree on the full set of species. However, wQFM, in general, can be used to amalgamate any set of given weighted quartets (e.g. dominant quartets or the quartets produced by SVDquartets, which we call wQFM-SVDquartets). Therefore, the broader impact and a clear benefit of wQFM over ASTRAL is that wQFM can be used outside the context of gene tree estimation. In this particular study, we have considered wQFM-GTF, where the frequency of a quartet in the input gene trees is used as a weight and so high weight suggests higher confidence in the quartet gene tree. However, weights can be inferred in other ways as well. For example, pairwise distances between taxa can be considered while computing the weights on quartets (Avni et al., 2015). The weights can be computed from input set of gene trees with or without generating bootstrapping gene tree samples [site-wise re-sampling, gene-wise re-sampling or both (Seo, 2008)]. wQFM uses a heuristic to combine the weighted quartet trees into a supertree, attempting to solve a version of the NP-hard ‘Maximum Quartet Compatibility’ problem (Jiang et al., 2001), where we set weights on the quartet trees.
2.4 Algorithmic pipeline of wQFM and wQFM-GTF
wQFM-GTF has two steps; in the first step, we generate a set of weighted quartet trees from the input set of gene trees, and in the second step, we estimate the species tree from the set of weighted quartet trees. We now describe how we perform each step. wQFM, in general, can take any set of given weighted quartets. Here, we discuss Step 1 in the context of wQFM-GTF.
2.4.1 Step 1: generate weighted quartets
Given a set of k gene trees on taxon set , we compute weights for every possible quartet tree of four leaves, where denotes the unrooted quartet tree with leaf set in which the pair a, b is separated from the pair c, d by an edge. Thus, we compute a weight w(q) for every possible (unrooted) quartet tree q. Note that on every set of four species, there are three possible unrooted quartet trees (simply called ‘quartets’). Also note that every gene tree on the set of taxa induces a single quartet tree on . We define the support for quartet tree to be the number of the trees in that induce on set .
2.4.2 Step 2: construct supertree
Our technique, which we call wQFM, to combine the quartet trees into a tree on the full set of taxa is the weighted version of the QFM technique, developed in Reaz et al. (2014). We briefly describe the algorithmic pipeline, and refer the readers to Reaz et al. (2014) for more details.
2.5 Terminology
For an unrooted tree T on taxon set , we let L(T) denote the leaf set of T. Every edge in T defines a bipartition of its leaf set (defined by deleting the edge but not its endpoints from T), which is denoted by πe. However, we can also refer to an arbitrary bipartition on set P, whether or not it is present in a given tree T; thus, we let (X, Y) be a bipartition with X on one side and Y on the other (note that the order of X and Y does not matter).
Under the assumption that all gene trees in the input are fully resolved, then given a bipartition (X, Y) on set , we partition the quartet trees defined by the input trees as follows:
quartet trees that are satisfied by (X, Y): those quartet trees where and , or and [i.e. the bipartition (X, Y) separates the two sibling leaf pairs with the quartet tree from each other],
quartet trees that are violated by (X, Y): those quartet trees q whose taxa are fully contained in , and where X and Y each contains exactly two of the four taxa in q but q is not satisfied by (X, Y) and
quartet trees that are deferred by (X, Y): those quartet trees q so that of its four taxa reside in X or in Y.
In fact, we can partition all possible quartet trees using any given bipartition, whether or not they appear in any input gene tree. We will refer to a pair (X, Y) with and as either a full bipartition (or simply a bipartition). A non-trivial bipartition is one that has at least two taxa on each side.
2.6 Divide-and-conquer approach
Let Q be a set of weighted quartet trees over a taxon set . The divide-and-conquer approach takes the pair (Q, P) as input. The basic divide-and-conquer algorithm operates in a top-down manner: a good non-trivial bipartition is produced, rooted trees are calculated on the two parts of the bipartition, and then combined together into a rooted tree on the full dataset by making them both subtrees of a common root. Then the tree is unrooted. The key to the algorithm is therefore finding the bipartition, and showing how to recurse on the subproblems so as to produce rooted trees.
This is the same basic top-down technique as used in QMC (Snir and Rao, 2010), so the only difference in the two methods is how the good non-trivial bipartition is produced. The differences in algorithm design define how the tree space is being searched and are important to the accuracy of the resultant tree.
We now briefly describe the technique used to find a good bipartition. We score a bipartition with respect to the set Q of quartet trees based on the difference between the total weight of all satisfied quartets and the total weight of all violated quartets. However, the partition score can be defined in other ways and the number of deferred quartets can be considered as well (Reaz et al., 2014). The technique to find the bipartition uses a heuristic iterative strategy, in which each iteration begins with the bipartition from the previous iteration, and tries to improve it. If the search strategy within this iteration finds a better bipartition, then a new iteration begins with the new bipartition. Thus, the strategy continues until it reaches a local optimum. The search within each iteration, however, allows for bipartitions with poorer scores to be computed, and hence the overall strategy is not purely hill-climbing. The running time of each iteration is polynomial, but the number of iterations depends on the search.
Given the final bipartition (A, B) on P, we use it to define two inputs to wQFM. By running wQFM recursively, we construct two rooted trees, one on A and one on B. We then create a rooted tree on (the full set of taxa), and then ignore the rooting to obtain an unrooted tree on . Thus, the rest of the algorithm depends on how we define these two inputs, and how we use wQFM to obtain rooted trees.
Letting denote the bipartition that is produced in the divide step, we divide Q into three sets, as follows. The first set contains all quartet trees that are either satisfied or violated by . The other two sets are QA and QB, where , and QB is defined similarly. Note that all quartet trees in QA and QB are deferred by .
For each quartet tree with , we label the taxon that is not in A by a new dummy taxon . We similarly relabel one leaf in the relevant quartet trees in QB with a new dummy taxon . This produces sets and , which are on sets and , respectively. We then recurse on each pair and , producing trees that we combine by identifying leaves and , and suppressing nodes of degree two. The base case is obtained when the taxon set has three or fewer leaves, in which case, we return a star. We do not pursue this further here, but see Reaz et al. (2014).
3 Experimental studies
3.1 Datasets
3.1.1 Simulated dataset
We studied previously used simulated and biological datasets to evaluate the performance of wQFM-GTF. We used two biologically-based simulated datasets (the avian and mammalian simulated datasets) studied in Mirarab et al. (2014c). We also analyzed three other simulated datasets (11-taxon, 15-taxon and 101-taxon) from Chung and Ané (2011), Mirarab et al. (2014c) and Mirarab and Warnow (2015). These datasets range from moderately low to extremely high levels of ILS, and range in terms of gene tree estimation errors and numbers of genes. Please see Supplementary Materials for details.
3.1.2 Biological dataset
We analyzed a collection of biological datasets: the 37-taxon mammalian dataset from Song et al. (2012), the avian phylogenomic dataset containing 48 species and 14 446 loci (including exons, introns and UCEs), the amniota dataset from Chiari et al. (2012) and the angiosperm dataset from Xi et al. (2014).
3.2 Methods
We compared wQFM-GTF with the best existing weighted quartet amalgamation method wQMC as well as with ASTRAL-III (Zhang et al., 2018) (version 5.7.3), which is considered as one of the most accurate and widely used coalescent-based species tree estimation methods. ASTRAL has been shown to outperform other coalescent-based methods (Chou et al., 2015; Davidson et al., 2015; Mirarab et al., 2014a), including MP-EST, BUCKy, NJst and SVDquartets. We ran wQFM-GTF and wQMC using the embedded quartets in the gene trees with weights reflecting the frequencies of the quartets. We use wQFM (rather than wQFM-GTF) in the rest of the manuscript for brevity and because the context of this study (i.e. species tree estimation from gene trees) is already clear.
3.3 Branch support estimation
We used a single ML tree estimate (we call this ‘best ML’ gene tree) for each gene to estimate species trees using ASTRAL, wQFM and wQMC. However, in order to draw branch supports on the estimated species trees, we used bootstrap ML gene trees for each gene. We used the site-only multi-locus bootstrapping procedure (Mirarab et al., 2014b) as follows. For each gene, 100 replicates of bootstrapping were performed. Next, 100 replicates () of input datasets to the summary methods are created such that Ri contains the i-th bootstrap tree across all genes with . ASTRAL, wQFM and wQMC were then run on these 100 replicates, and 100 species trees were estimated. Finally, support values were drawn on the branches of the trees estimated on best ML gene trees by counting the occurrences of each bipartition in the 100 species trees. See Seo (2008) for a discussion of more elaborate approaches for bootstrapping multi-locus datasets, including re-sampling both sites and genes. We also assessed support values in the estimated trees using local posterior probabilities (Sayyari and Mirarab, 2016) computed by ASTRAL. The local posterior probabilities are computed based on a transformation of normalized quartet scores (the percentage of quartets in the gene trees that agree with a branch in the estimated species tree).
3.4 Measurements
We compared the estimated trees (on simulated datasets) with the model species tree using normalized Robinson–Foulds (RF) distance (Robinson and Foulds, 1981) to measure the tree error. The RF distance between two trees is the sum of the bipartitions (splits) induced by one tree but not by the other, and vice versa. We also compared the quartet scores of the trees estimated by different methods. All the trees estimated in this study are binary and so false positive and false negative and RF rates are identical. For the biological dataset, we compared the estimated species trees to the scientific literature. We analyzed multiple replicates of data for various model conditions and performed two-sided Wilcoxon signed-rank test (with ) to measure the statistical significance of the differences between two methods.
4 Results and discussion
4.1 Results on 37-taxon dataset
The average RF rates of wQFM, wQMC and ASTRAL on various model conditions in 37-taxon dataset are shown in Figure 1. Overall, ASTRAL, wQFM and wQMC had competitive accuracy, but wQFM achieved higher tree accuracy than both ASTRAL and wQMC on most of the model conditions (albeit the differences are small). The impact of changes in the ILS level with 500 bp sequences and 200 genes is shown in Figure 1a. As expected, species tree error rates increased as ILS levels increased. ASTRAL and wQMC had similar accuracy, and wQFM was slightly better than these two existing methods. Figure 1b shows the impact of varying amounts of gene tree estimation error (controlled by sequence lengths). All methods showed improved accuracy as the sequence length was increased, and best results were obtained on true gene trees. wQFM consistently produced slightly better species trees than ASTRAL and wQMC, and its improvement over ASTRAL and wQMC is statistically significant on the 250bp model condition. Figure 1c shows that all of these methods improved as we increased the number of genes. wQFM matched the accuracy of ASTRAL and wQMC with a slight advantage on model conditions with relatively larger numbers of genes.
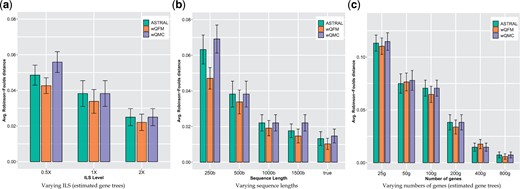
Comparison of ASTRAL, wQFM and wQMC on 37-taxon simulated mammalian dataset. We show the average RF rates with standard error bars over 20 replicates. (a) The level of ILS was varied from 0.5X (highest) to 2X (lowest) amount, keeping the sequence length fixed at 500 bp and the number of genes at 200. (b) The sequence length was varied from 250 bp to 1500 bp, keeping the number of genes fixed at 200, and ILS at 1X (moderate ILS). We also analyzed the true gene trees. (c) The number of genes was varied from 25 to 800, with 500 bp sequence length and moderate (1X) ILS
4.2 Results on 48-taxon avian simulated dataset
Figure 2 shows the performance of various methods on: (i) varying ILS levels (0.5X, 1X and 2X) with 1000 genes and a fixed sequence length (500 bp), and (ii) varying numbers of genes with a fixed level of ILS (1X level) and 500 bp sequence length. We analyzed both estimated and true gene trees. Across all the model conditions, wQFM outperformed both ASTRAL and WQMC and, in many cases, the improvements are notable and statistically significant (). Unlike 37-taxon dataset, wQMC was substantially worse than wQFM and ASTRAL. Decreasing the estimation error (by increasing the sequence lengths), increasing the numbers of genes and using true gene trees (with no estimation error) improved the tree accuracy of all of these methods.
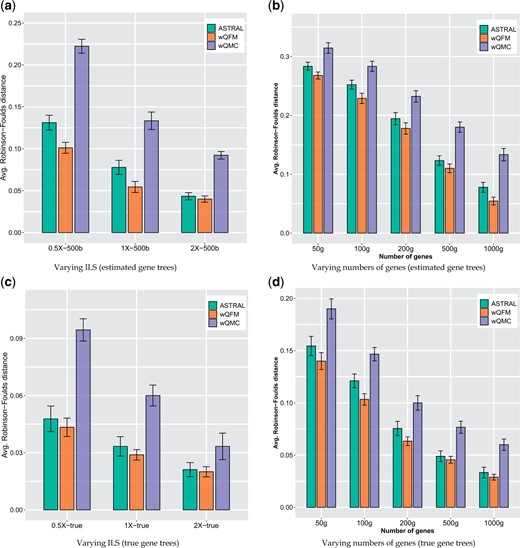
Comparison of ASTRAL, wQFM and wQMC on 48-taxon simulated avian dataset. We show the average RF rates with standard error bars over 20 replicates. (a) The level of ILS was varied from 0.5X (highest) to 2X (lowest) amount, keeping the sequence length fixed at 500 bp and the number of genes at 1000, (b) number of genes was varied from 50 to 1000 g, with 500 bp sequence length and moderate (1X) ILS. (c and d) Performance on true gene trees with varying levels of ILS and varying numbers of genes
4.3 Results on 15-taxon dataset
We have investigated the performance on varying gene tree estimation errors using 100 and 1000 bp sequence lengths and on varying numbers of genes (100 and 1000) as shown in Figure 3. wQFM achieved better tree accuracy than ASTRAL and wQMC on all the model conditions with estimated gene trees. The accuracy of ASTRAl and wQMC was comparable. All of these methods improved as the number of genes and the length of the sequence increased, and they produced the best results (RF rate ) on true gene trees.
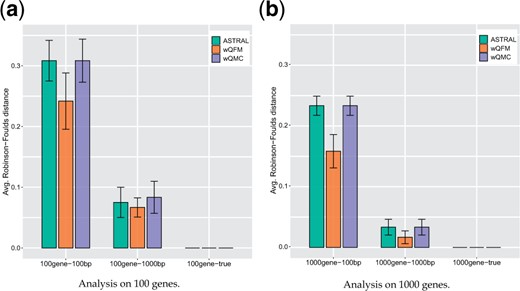
Comparison of ASTRAL, wQFM and wQMC on 15-taxon dataset. We show the average RF rates with standard errors over 10 replicates. We analyzed both estimated gene trees (with different amounts of gene tree estimation error, controlled by sequence lengths) and true gene trees
4.4 Results on 11-taxon and 101-taxon datasets
Similar trends were observed on 11-taxon and 101-taxon datasets (see Supplementary Section 3).
4.5 Quartet scores
We compared the quartet scores of the trees estimated by wQFM, ASTRAL and wQMC to assess the performance of these methods in terms of solving the problem. The quartet scores of these methods on different datasets (shown in Supplementary Tables S1–S6) suggest that ASTRAL is better than or as good as wQFM and wQMC in solving the problem. However, in many cases, the differences are small.
Although maximizing quartet score is a statistically consistent approach for estimating species trees, the statistical guarantee does not hold when we have a limited number of gene trees with estimation error, which is often so for practical phylogenomic datasets. In the presence of small numbers of estimated gene trees, summary methods may ‘overshoot’ the optimization criteria, meaning that the optimal solution to the problem is not the true species tree [see the quartet scores of the model species trees and the estimated species trees in Supplementary Tables S1–S6, and also Farah et al. (2020) for more relevant discussions]. Although ASTRAL achieves matching or higher quartet scores than wQFM, ASTRAL produced less accurate trees than wQFM in many cases. More interestingly, the quartet scores of wQFM and wQMC are comparable, but the tree accuracy of wQFM is significantly better than wQMC on most of the datasets analyzed in this study (see Figs 1–3 and Supplementary Tables S1–S4). We believe that the differences in algorithm design define how the tree space is being searched and are important to the accuracy of the resultant tree, especially when we have a limited number of estimated gene trees (with estimation error).
4.6 Results on biological dataset
4.6.1 Avian dataset
We have re-analyzed the avian biological dataset containing 14 446 loci across 48 taxa (including exons, introns and UCEs). We have also analyzed 8251 exon, 2516 intron and 3679 UCE gene trees separately. This is an extremely challenging dataset since it contains high levels of gene tree discord, perhaps because their ancestors experienced a rapid radiation (Jarvis et al., 2014). The original analyses of this data by MP-EST using the binned gene trees was largely congruent with combined analyses using ExaML (Kozlov et al., 2015), and both trees were presented as reference (Jarvis et al., 2014; Mirarab et al., 2014c).
Similar to the unbinned MP-EST analysis (Bayzid et al., 2015; Jarvis et al., 2014; Mirarab et al., 2014c), ASTRAL, wQFM and wQMC run on 14 446 unbinned gene trees violate several sub-groups established in the avian phylogenomics project and other studies (see Fig. 4). However, wQFM tree is closer to the MP-EST tree (on binned gene trees) than the trees estimated by ASTRAL and wQMC.
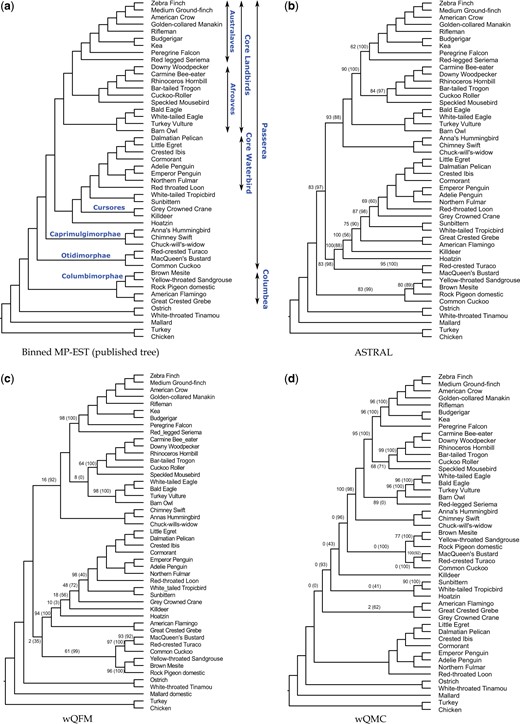
Estimated trees on the avian dataset with 14 446 genes. (a) Reference trees from the original paper (Jarvis et al., 2014) using MP-EST with statistical binning (Mirarab et al., 2014c), (b–d) trees estimated by ASTRAL, wQFM and wQMC, respectively, on 14 446 unbinned gene trees. Two branch support (BS) values are shown: the first value (without parentheses) is based on site-only MLBS and the second value (with parentheses) is quartet-based local posterior probability (multiplied by 100). All BS values are 100% except where noted
Both wQFM and ASTRAL correctly placed seriemas as sister to the clade containing passerimorphae (passeriformes and parrot) and falcon, and thus reconstructed the well-established Australaves clade (passeriformes, parrot, falcon and seriema) with high support. However, wQMC misplaced seriemas and failed to recover Australaves. ASTRAL and wQFM correctly reconstructed the core landbirds and core waterbirds (Cracraft, 2013; Ericson et al., 2006; Hackett et al., 2008; Jarvis et al., 2014; Kimball et al., 2013). wQMC also grouped them together, albeit with different branching orderings. While these methods recovered the core landbirds and core waterbirds (Cracraft, 2013; Ericson et al., 2006; Hackett et al., 2008; Jarvis et al., 2014; Kimball et al., 2013) as well as various smaller sub-groups (e.g. Passerimorphae, Accipitrimorphae, Phaethontimorphae, Caprimulgimorphae and Phoenicopterimorphae), they could not recover some key clades. All of them failed to recover Columbea (flamingo, grebe, pigeon, mesite and sandgrouse). Although all of them correctly constructed Columbimorphae (mesite, sandgrouse and pigeon) and Phoenicopterimorphae (flamingo and grebe) with reasonably high support, they did not place them as sister clades and thus failed to recover Columbea. ASTRAL failed to recover Otidimorphae (bustard, turaco and cuckoo), whereas both wQFM and wQMC reconstructed this clade. All of these methods failed to recover Cursores (crane and killdeer).
The phylogeny of Neoaves has remained intractable despite large-scale efforts using genome-wide data (Hackett et al., 2008; Jarvis et al., 2014; Kimball et al., 2009; McCormack et al., 2013; Prum et al., 2015). There are notable topological differences among trees produced in different phylogenomic studies. In addition to the difficulties resulting from the rapid radiation of this group of birds, ‘data-type effect’ (i.e. different data types have distinct sets of estimated gene trees) contributes to this conflict (Braun and Kimball, 2021; Reddy et al., 2017). Therefore, we assessed the performance of wQFM, ASTRAL and wQFM on exon, intron and UCE gene trees separately. Our analyses support previous findings as the trees estimated on different sets of gene trees exhibit a number of conflicts (see Supplementary Fig. S3).
4.6.2 Amniota dataset
We re-analyzed the amniota dataset [both amino acid (AA) and nucleotide (DNA) gene trees] from Chiari et al. (2012) containing 248 genes across 16 amniota taxa. The goal is to resolve the position of turtles relative to birds and crocodiles. Previous studies (Chiari et al., 2012; Hugall et al., 2007; Iwabe et al., 2005; Mirarab et al., 2014b) suggest the sister relationship between birds and crocodiles (forming archosaurs), and the placement of turtles as the sister to archosaurs.
ASTRAL, wQMC and wQFM, either on AA or DNA gene trees, correctly put turtles as a sister clade to archosaurs with high support (see Fig. 5). All these three methods, either on AA or DNA data, reconstructed identical trees and these two trees (on AA and DNA data) are highly congruent, except for the resolution within the turtles (Phrynops hilarii, Caretta caretta, Chelonoidis nigra and Emys orbicularis).
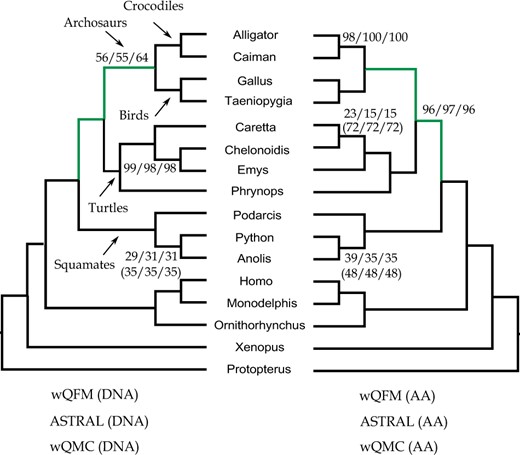
Analyses of the amniota dataset using ASTRAL, wQFM and wQMC. We analyzed both DNA and AA gene trees. Two branch support values are shown: the values without parentheses are based on site-only MLBS and the values with parentheses are quartet-based local posterior probabilities (multiplied by 100). For each of these two types of support, we show the BS for three methods (wQFM, ASTRAL and wQMC) in the following order: wQFM/ASTRAL/wQMC. All BS values are 100% except where noted
4.6.3 Mammalian dataset
We re-analyzed the mammalian dataset from Song et al. (2012) containing 447 genes across 37 mammals after removing 21 mislabeled genes (confirmed by the authors), and two other outlier genes. The trees produced by wQFM, wQMC and ASTRAL are identical to each other (see Supplementary Fig. S4). This tree placed tree shrews (Tupaia belangeri) as sister to Glires with high support, which is consistent to the CA-ML analyses [reported in Song et al. (2012)], and bats have been placed as sister to the clade containing Cetartiodactyla, Carnivora and Perissodactyla [which is consistent to the MP-EST analyses (Mirarab et al., 2014b)]. However, alternative relationships (e.g. tree shrew as sister to Primates, and bats as sister to Cetartiodactyla) have also been observed (Mirarab et al., 2014a, b). The placement of tree shrews and bats is of substantial debate (Boussau et al., 2013; Janečka et al., 2007; Jingyang et al., 2012; Kumar et al., 2013), and so the differential placement is of considerable interest in mammalian systematics.
4.6.4 Angiosperm dataset
We analyzed the angriosperm dataset from Xi et al. (2014) containing 310 genes samples from 42 angiosperms and 4 outgroups. The key question here is to investigate the position of Amborella trichopoda Baill. We provide the trees estimated by different methods in the Supplementary Materials.
4.7 Comparison with QFM: impact of using weighted quartets
Both QFM and wQFM construct trees from a collection of quartets, but wQFM is capable of handling weighted quartets. One of the motivations for using the weighted version is to use all possible quartets [i.e. quartets for n taxa] with relative weights instead of using the unweighted setting, where—for each set of four taxa—we are given with one of the three alternate quartet topologies (Chifman and Kubatko, 2014; Reaz et al., 2014). In order to show the efficacy of using weighted quartets, we have compared wQFM with QFM (see Supplementary Section 4). Experimental results suggest that assigning weights can improve phylogenomic analysis.
4.8 Running time
On smaller datasets with limited numbers of taxa and genes (e.g. 15-, 37- and 48-taxon simulated datasets), ASTRAL, wQFM and wQMC are reasonably fast (take only a few seconds for most of the model conditions) and there is no substantial differences in running time. However, as we increase the numbers of taxa and genes, notable differences in running time were observed. For example, ASTRAL took around 32 h to run on the avian biological dataset with 14K gene trees. wQMC and wQFM, on the other hand, finished within 2 and 20 s, respectively. This is due to the fact that ASTRAL’s running time increases as we increase the number of genes (hence the number of unique bipartitions), but wQMC and wQFM takes as input a set of weighted quartets and thus their running times are not sensitive to the number of genes. On a relatively larger dataset with 101 taxa, the running time of wQFM ranges from 25 to 40 min. ASTRAL and wQMC were much faster, taking around 2–3 min and 5 s, respectively. See Supplementary Materials for more information about running times under different model conditions.
5 Conclusions
We present wQFM—a new fast and highly accurate method for estimating species trees from genome-scale data by amalgamating weighted quartets, which matches or improves upon the best existing methods under a range of realistic model conditions. Quartet amalgamation is an important class of methods, which takes individual quartets as input and amalgamates them into a single coherent tree. With the recent advances in computing accurate quartet estimation using site pattern probabilities without needing to estimate gene trees (Chifman and Kubatko, 2014), and thereby reducing the impact of gene tree estimation error in species tree estimation, quartet amalgamation techniques like QFM are being widely used and have drawn substantial attention from the systematists. Moreover, assigning relative weights to the quartets can potentially improve the tree accuracy (Avni et al., 2015), and thus can be more applicable than weight-oblivious techniques like QMC and QFM for estimating species trees from multi-locus data.
wQFM is a new divide-and-conquer based method for amalgamating weighted quartets. Our wide-ranging experimental results suggest that wQFM can reliably estimate species trees under practical and challenging model conditions. We showed that wQFM can estimate trees with similar or better accuracy than the main two alternate methods, wQMC and ASTRAL. Thus, wQFM advances the state-of-the-art in weighted quartet amalgamation and can be used to accurately analyze upcoming multi-gene datasets. However, this study can be extended in several directions. This study is limited to small to moderate size datasets. Future studies need to investigate the performance of wQFM on relatively larger phylogenomic datasets with hundreds of taxa. This study was limited in terms of the weights of the quartets that were explored, in that we restricted the analysis to weights generated using the frequency of the quartets in the input gene trees. However, to demonstrate the broader utility of wQFM, namely that pipelines using wQFM are not required to have gene trees as input (which is the case for ASTRAL), future studies need to investigate the performance of wQFM on weighted quartets generated by different approaches (e.g. SVDquartets). wQFM has been evaluated on various datasets simulated under ILS. However, evaluating the performance of wQFM (weighted quartet amalgamation techniques in general) in the presence of horizontal gene transfer and gene duplications and losses would be interesting, especially given recent findings that the dominant quartet agrees with the species tree under some models of gene duplication and loss (Legried et al., 2020) and some models of horizontal gene transfer (Roch and Snir, 2013). On an ending note, this study shows that the idea of estimating species trees by amalgamating weighted quartets has merit and should be pursued and used in future phylogenomic studies.
Funding
This work was partially supported by the Information and Communication Technology Division (ICT Division), Government of the People’s Republic of Bangladesh.
Conflict of Interest: none declared.
References
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}