





Abstract
CONSTAX—the CONSensus TAXonomy classifier—was developed for accurate and reproducible taxonomic annotation of fungal rDNA amplicon sequences and is based upon a consensus approach of RDP, SINTAX and UTAX algorithms. CONSTAX2 extends these features to classify prokaryotes as well as eukaryotes and incorporates BLAST-based classifiers to reduce classification errors. Additionally, CONSTAX2 implements a conda-installable command-line tool with improved classification metrics, faster training, multithreading support, capacity to incorporate external taxonomic databases and new isolate matching and high-level taxonomy tools, replete with documentation and example tutorials.
CONSTAX2 is available at https://github.com/liberjul/CONSTAXv2, and is packaged for Linux and MacOS from Bioconda with use under the MIT License. A tutorial and documentation are available at https://constax.readthedocs.io/en/latest/. Data and scripts associated with the manuscript are available at https://github.com/liberjul/CONSTAXv2_ms_code.
Supplementary data are available at Bioinformatics online.
1 Introduction
High-throughput sequencing has revolutionized metagenomics and microbiome sciences (Di Bella et al., 2013). These culture-independent methods have revealed previously unrecognized microbial diversity and has allowed researchers to detect organisms occurring at extremely low abundances (Brown et al., 2015). Amplicon-based sequencing, which relies on amplification and sequencing of genetic markers such as the rRNA operon or protein-coding genes, is an extremely popular technique for studying microbiomes and microbial communities. Following sequencing, quality control and demultiplexing, amplicon reads are clustered and representative sequences are classified taxonomically. Many algorithms have been developed to conduct the task of assigning taxonomy to environmental sequences. Some of the most popular include BLAST-based tools (Altschul et al., 1997; Bokulich et al., 2018), the Ribosomal Database Project (RDP) naive Bayesian classifier (Wang et al., 2007) and the USEARCH algorithms SINTAX (Edgar, 2016) and UTAX (Edgar, 2013).
While each of these tools can be implemented independently to assign taxonomy, a consensus-based approach was demonstrated to increase the accuracy and number of sequences with taxonomic assignments (Gdanetz et al., 2017). Since the original release of the CONSTAX (CONSensus TAXonomy) classifier, we have found the need for improved ease of use, updated software compatibility, simpler installation, improved accuracy and adaptability and application to bacteria and other organisms. To address these needs, an updated version, CONSTAX2, has been developed.
2 Implementation
CONSTAX2 (referred to hereafter as ‘CONSTAX’) is released as a conda-installable command-line tool, available from the bioconda installation channel (Grüning et al., 2018) for Linux, MacOS and WSL systems. It is installed with the command ‘conda install -c bioconda constax’, see https://github.com/liberjul/CONSTAXv2. CONSTAX requires two files: (1) ‘-d, –db’ a reference database file in FASTA format (Pearson and Lipman, 1988) with header lines containing taxonomy of the sequences in the SILVA (Glöckner et al., 2017) or UNITE (Nilsson et al., 2019) format and (2) ‘-i, –input’ an input file of user-submitted query sequences in the FASTA format. This version implements a BLAST classification algorithm instead of the legacy UTAX classifier if the ‘-b, –blast’ flag is used.
The user may designate several additional parameters, including confidence threshold for assignment (‘-c, –conf’), BLAST classifier parameters, and whether to use a conservative consensus rule (‘–conservative’), which requires agreement of two (instead of one) nonnull assignments to assign a taxonomy at the given rank. CONSTAX offers multithreaded classification with the argument, ‘-n, –num_threads’.
CONSTAX generates three directories while running: (1) training files directory (‘-f, –trainfile’), (2) taxonomy assignments directory (‘-x, –tax’) and (3) an output directory (‘-o, –output’). Prior to classifying sequences, training must be performed on any newly used database file with the ‘-t, –train’ flag. After initial training, generated training files can be used in any later run by specifying the same training files directory. When training is performed, CONSTAX will automatically generate the formatted database files required by each classifier, as long as the supplied database has SILVA or UNITE header formatting. Following training, the classification or search command is performed for each classifier, and files are output to the taxonomic assignments directory. Finally, each classification output is reformatted and used to generate a consensus hierarchical taxonomy, then each classifier’s result and the consensus result are stored in the output directory as tab-delimited value files with each row corresponding to a query sequence and values as the hierarchical taxonomy assigned to each query.
CONSTAX2 offers two additional features: (1) the ability to match input sequences to isolates using the ‘–isolates’ option and (2) the ability to determine higher-level taxonomy using representative databases with the ‘–high_level_db’ option. Both approaches implement the BLAST algorithm to associate input sequences with hits from the respective databases, returning a single best hit with a default threshold of query cover ≥75% and E value ≤10. Cutoffs for query coverage and percent identity can be specified. Isolate matching is designed to find best matches to sequenced organisms in pure culture or any known reference sequences, which may streamline culture-dependent and culture-independent analyses, and can also be used to implicate potential contamination by association with known isolates previously worked with in the laboratory or sequencing facility where the samples were processed. Higher-level taxonomy designations are also useful in filtering host, organelles or nontarget taxa, which may show up in rDNA surveys. For 16S rDNA prokaryote datasets, the latest SILVA SSURef NR99 database is recommended, while the latest UNITE Eukaryotes database is recommended for ITS studies of Fungi.
3 Results
3.1 Algorithm speed and memory usage
The implementation of the BLAST algorithm as a third classifier and replacement of UTAX provides crucial speedup of the training step (Fig. 1A), facilitating the use of the much larger SILVA database. For 16 000 sequences randomly sampled from the SILVA database, the BLAST implementation (including SINTAX, RDP and BLAST) trained 370 ± 32.1 sequences * s−1 (mean ± SD), while the UTAX implementation (including SINTAX, RDP and UTAX) trained 41.9 ± 0.911 sequences * s−1, an ∼9-fold improvement. Furthermore, the BLAST implementation trains faster per sequence at larger database sizes.
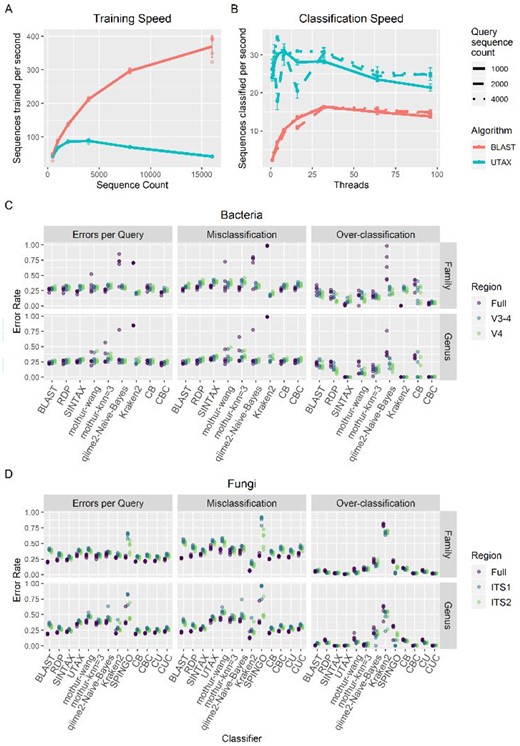
Performance of the CONSTAX algorithm. (A) Reference sequences parsed per second for training of the CONSTAX implementation with BLAST and UTAX, as a function of the size of the training set. (B) Sequences classified per second with BLAST and UTAX implementations, as a function of query set size and threads used for parallelization. (C and D) Classification performance resulting from clade-partition cross-validation, at genus and family partition ranks, for full and extracted regions, corresponding to each CONSTAX classifier and other common classification tools, for (C) bacteria in the SILVA SSURef release 138 dataset and (D) fungi in the UNITE RepS February 4, 2020 general release. EPQ, misclassification rate and over-classification rate are defined by Edgar (2016) and in Supplementary Data. CB—CONSTAX with BLAST, CBC—CONSTAX with BLAST and conservative rule, CU—CONSTAX with UTAX, CUC—CONSTAX with UTAX and conservative rule
Although the BLAST implementation is faster for training, classification is faster with the UTAX implementation (Fig. 1B). The maximum classification speed was achieved at 32 threads for the BLAST implementation and between 4 and 8 threads for the UTAX implementation, depending on the number of query sequences classified, which minorly affected per-sequence rates. At 4000 query sequences, the BLAST implementation classified at a speed of 16.3 ± 0.298 sequences * s−1 on 32 threads, while the UTAX implementation classified at a speed of 34.4 ± 0.611 sequences * s−1 on 4 threads.
Training with bacterial records in the SILVA 138 SSU release (1 983 818 sequences, 2.8 Gb) with the BLAST implementation used 102.96 GB of RAM, while the fungal UNITE database (95 481 sequences, 60 Mb) used 15.24 GB for BLAST and 12.72 GB for UTAX implementations. Classification with the SILVA database with 16 threads used 28.16 GB for 500 sequences and 30.88 GB for 1000 sequences, while the UNITE database used 6–7 GB, regardless of implementation, threads or number of query sequences.
3.2 Algorithm performance
Clade partitioned cross-validation and classification metrics from SINTAX (Edgar, 2016) were used (Supplementary Data) on the taxonomy assignments of each classifier and the consensus, which were compared for genus and family-level partitions as well as for full length ITS1-5.8S-ITS2 or 16S regions (accounting for the commonly used subregions ITS1, ITS2, V4 and V3–4) with errors per query (EPQ; sum of false negative and false positive rates), over-classification (false positive rate of unknown taxa) and misclassification (false positive rate of known taxa), for five query-reference paired datasets (Fig. 1C and D, Supplementary Table S1). The popular mothur knn and Wang classifiers (Schloss et al., 2009), qiime q2-feature-classifier plugin (Bokulich et al., 2018), Kraken 2 (Wood et al., 2019) and SPINGO (Allard et al., 2015) classifiers were compared using the same protocol. CONSTAX with the nonconservative consensus with BLAST had the fewest EPQ for any classifier (0.236–0.248, 95% CI for all regions and partition levels), or tied for fewest with the UTAX consensus, across the UNITE dataset. Alternatively, CONSTAX with the conservative consensus with BLAST had the fewest errors for all classifications in the SILVA dataset (EPQ = 0.214–0.259). The BLAST implementation was valuable in decreasing misclassifications for the UNITE dataset compared to UTAX, but this was generally associated with increased (erroneous) over-classifications.
4 Conclusion
The newest implementation of CONSTAX offers improvement over its predecessor by ease of use and improved applicability and accuracy. Hierarchical taxonomy classification accuracy by a consensus approach in CONSTAX2 is demonstrated to outperform commonly used classifiers while remaining computationally feasible.
Acknowledgements
The authors thank Zachary Noel, Reid Longley, Acer VanWallendael and Shay Shemanski for helping test the software. We thank Natalie Vande Pol for assistance in the version transition.
Funding
This work has been supported by the US National Science Foundation DEB 1737898 to G.B. and J.L. and through the Great Lakes Bioenergy Research Center, U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, under award number DE-SC0018409 to G.B. and G.M.N.B.
Conflict of Interest: none declared.


{kind=link}