





Abstract
Contact predictions within a protein have recently become a viable method for accurate prediction of protein structure. Using predicted distance distributions has been shown in many cases to be superior to only using a binary contact annotation. Using predicted interprotein distances has also been shown to be able to dock some protein dimers.
Here, we present pyconsFold. Using CNS as its underlying folding mechanism and predicted contact distance it outperforms regular contact prediction-based modeling on our dataset of 210 proteins. It performs marginally worse than the state-of-the-art pyRosetta folding pipeline but is on average about 20 times faster per model. More importantly pyconsFold can also be used as a fold-and-dock protocol by using predicted interprotein contacts/distances to simultaneously fold and dock two protein chains.
pyconsFold is implemented in Python 3 with a strong focus on using as few dependencies as possible for longevity. It is available both as a pip package in Python 3 and as source code on GitHub and is published under the GPLv3 license. The data underlying this article together with source code are available on github, at https://github.com/johnlamb/pyconsfold.
Supplementary data are available at Bioinformatics online.
1 pyconsFold
De novo protein modeling has recently seen significant improvements by relying on contact predictions that have been presented in binary format, two residues are either in contact or not. However, today the best methods are leveraging distance predictions (CASP13; Kryshtafovych et al., 2019; Yang et al., 2020) providing a higher accuracy of the models than if binary contacts were used. To generate a model, it is necessary to feed the contact/distance maps into a modeling program. One of the most popular approaches is CONFOLD (Adhikari et al., 2015), which is a wrapper around CNS (Brunger, 2007), that uses predicted binary contacts together with predicted secondary structure to model proteins. Here, we introduce pyconsFold, a reimplementation and extension of CONFOLD that achieves better results using distance predictions and that also expands to allow for more geometric restraints, such as angles predicted by tools such as trRosetta (Yang et al., 2020). Finally, pyconsFold introduces the first easily accessible method for fold-and-dock of two protein chains from interchain contacts.
2 Modeling
pyconsFold uses predicted distance between pairs of amino acid residues in a sequence. These distances together with either predicted or fixed errors are translated into geometric constraints that are used together with CNS to model the full protein structure. pyconsFold can also be run in contact mode which simulates using binary contact predictions without a predicted distance, this is basically identical to CONFOLD. If side chain angles, for instance predicted by trRosetta, are present, they can also be used as input for further geometric constraint. We have, however, not seen any significant improvement in the model quality using the angles as constraints.
In Supplementary Figure S1 and Table S1, the TMscores of pyconsFold generated models compared with models from CONFOLD (Adhikari et al., 2015) and trRosetta (Yang et al., 2020) on the PconsC3-dataset (Michel et al., 2017) (Supplementary Fig. S1) and CASP13 models (Supplementary Table S1). The results show that distance-based modeling outperforms contact-based modeling in almost all cases. Distance prediction outperforms binary contact predictions and pyconsFold achieves comparable results on most target with pyRosetta.
3 Docking
pyconsFold introduces a new way of de novo docking together with folding. By using contact predictions which contains both inter- and intraresidue contacts, both folding and docking can be done simultaneously. Distance predictions of this type can be done by horizontally concatenating two multiple sequence alignments (MSAs) from two different chains in a complex and adding a poly-G region in between. The poly-G region prevents any spurious false predictions between the end of the first chain and the beginning of the second solely based on proximity. The poly-G region would be trimmed away and residues renumbered before the input is ready for pyconsFold.
As can be seen in Figure 1, using distances from the structure produces both very good docking and individual models. Replacing the interchain distances with predicted contacts significantly lowers the DockQ-score (Basu and Wallner, 2016), but the individual models TMscore does not decrease to the same extent, indicating that interchain contact predictions are less accurate. However, a full study of this is beyond the goals of this paper.
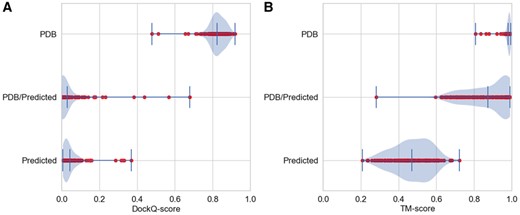
Performance of pyconsFold docking benchmarked on all 222 heterodimeric pairs from Dockground 4.3 (Kundrotas et al., 2020); PDB: real distances from PDB-file; PDB/Predicted: pdb-distance for intrachain distances and predicted interchain distances; Predicted: predicted intra- and interchain distances. (A) DockQ-score and (B) average TMscore of both chains
The pyconsFold docking protocol can be used in multiple ways. The inter- and intercontacts needed can be obtain from different sources and combined. This opens up for hybrid methods, e.g. where one dimer has a structural homolog from which distances can be extracted and combined with predicted distances for the other dimer and the interchain contacts.
4 Additional features
For ease of use and reproducibility, we have included several extra features and utilities. By default the generated models are ranked by CNS internal NOE energy, but Quality Assessment score pcons (Wallner and Elofsson, 2005) will also be calculated. If a native structure is known and supplied with the tmscore_pdb_file argument, the TMscore (Xu and Zhang, 2010; Zhang and Skolnick, 2007) for each model against the native structure will be calculated. Compiled versions of both Pcons and TMscore for unix based ×64 systems are packaged together with pyconsFold under the open-source Boost license. If your system does not support the built in versions, you can manually install them on your system and as long as they are in your path, will be chosen instead of the built in binaries.
Multiple utility functions are also included to make the extraction and conversion of distances and contacts from structure pdb/mmcif-files possible, see ‘Extras’ in the github repository.
5 Conclusion
pyconsFold offers a complete toolkit for de novo modeling using predicted contact distances and angles. Its focus is on ease of use and reproducibility and is available both as source on github and as an easily installable pip package in Python 3. It comes packaged with QA-programs to rank the generated models and allows transparency of parameters for the underlying CNS-system. Multiple test cases and examples are available in the github repository to demonstrate both advanced parameters and additional features. It also offers an innovative de novo fold-and-dock protocol where predicted interchain contacts are used as restraints for docking. This docking protocol is highly flexible and allows inputs to be a combination from structure and prediction based on the available material. It offers a significant increase in model accuracy over contact-based protocols by using predicted distances, see Supplementary Figure S1, and a comparable performance to pyRosetta although being around 20 times faster.
Funding
This work was supported by grants from the Swedish Research Council (VR-NT 2016-03798) and SNIC to A.E.
Conflict of Interest: none declared.


{kind=link}