





Abstract
The use and functionality of Electronic Health Records (EHR) have increased rapidly in the past few decades. EHRs are becoming an important depository of patient health information and can capture family data. Pedigree analysis is a longstanding and powerful approach that can gain insight into the underlying genetic and environmental factors in human health, but traditional approaches to identifying and recruiting families are low-throughput and labor-intensive. Therefore, high-throughput methods to automatically construct family pedigrees are needed.
We developed a stand-alone application: Electronic Pedigrees, or E-Pedigrees, which combines two validated family prediction algorithms into a single software package for high throughput pedigrees construction. The convenient platform considers patients’ basic demographic information and/or emergency contact data to infer high-accuracy parent–child relationship. Importantly, E-Pedigrees allows users to layer in additional pedigree data when available and provides options for applying different logical rules to improve accuracy of inferred family relationships. This software is fast and easy to use, is compatible with different EHR data sources, and its output is a standard PED file appropriate for multiple downstream analyses.
The Python 3.3+ version E-Pedigrees application is freely available on: https://github.com/xiayuan-huang/E-pedigrees.
1 Introduction
Family pedigrees are constructed as a set of directed acyclic graphs. Each node in the graph represents an individual in a family unit and the edges imply the social and genetic relationships. When layered with phenotypic information, pedigree data can provide important insights into human disease with distinct advantages over population-based studies of unrelated individuals (Hebbring, 2019; Huang et al., 2018; Leiser et al., 2002). Even data acquired from small pedigrees (e.g. trios) still have great value in genetic and epidemiologic research (Chen et al., 2013; Teo et al., 2009; Toptas et al., 2018). Unfortunately, accumulating informative family pedigrees can be costly to identify and recruit, and can be further challenged by incomplete and biased ascertainment.
An expanding source for rich phenotypic data collected longitudinally and in real time is an electronic health record (EHR). EHR systems are being used to predict disease risk for clinical care (Goldstein et al., 2017; Wang et al., 2014) and applied for genetic research. This is exemplified by a growing number of biobanks that link genetic information to EHR data such as the All of Us Research Program (https://allofus.nih.gov/), which facilitates its important applications in Mendelian diseases and population genomics (Garg et al., 2020). However, most EHR systems do not document a patient’s parental lineage or other familial relationships in a structured format.
In light of the growing influence of EHR systems on clinical care and advantages of family-based study designs in research, we developed a novel fully automated application called Electronic Pedigrees (E-Pedigrees) to construct family pedigrees from information readily available in an EHR system. It infers familial relationships using two previously published prediction algorithms including Family Pedigree Prediction Algorithm (FPPA) (Huang et al., 2018) and Relationship Inference from the Electronic Health Record (RIFTEHR) (Polubriaginof et al., 2018). FPPA algorithm predicts parent–child relationships from basic demographic data in the EHR, whereas RIFTEHR can capture other familial relationships using self-reported data in an EHR (i.e. emergency contact information). With the cooperation of these two algorithms together, more accurate and complete families can be identified. To ensure E-Pedigrees remains flexible and relevant as new data sources become available in the future, users can layer additional family data from tertiary sources (e.g. hospital birth records, public records or biobank data) and select different rule-based matching processes. The output is a standardized PED file with no contradictions applicable for many downstream pedigree-based analyses (Kaplanis et al., 2018; Liu et al., 2017; Shor et al., 2019). Compared to our previous developed algorithms (Huang et al., 2018; Polubriaginof et al., 2018), our newly developed application resolves a few limitations. (i) It is more adaptable to different EHR data sources. (ii) E-Pedigrees can generate larger and more complete families. (iii) The software can predict more family relationships with higher accuracy. (iv) E-Pedigrees has the capacity to capture unique family structures including half-siblings. (v) And the software can track family structures as new couplings occur over time.
2 Materials and methods
Figure 1 shows the input/output and the flowchart of the processes. User only needs to provide input data at the beginning; the application will automatically run all steps and result in a standardized PED file.
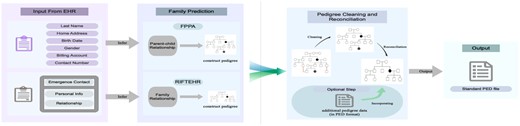
Flowchart for the E-pedigree application. This flowchart shows a parallel process of pedigree inference with two algorithms. In the ‘Pedigree Cleaning and Reconciliation’ step, E-pedigree combines and reconciliates the resulting pedigrees, and with the option of incorporating additional pedigree data, it outputs a standard PED file with predicted pedigrees
2.1 Family relationship references
Our E-Pedigrees application requires users to provide standard input of basic demographic information, such as last name, home address, date of birth, gender, billing account and contact number. Another input variable that can be included is emergency contact data, which includes the person’s name, address, phone number and their relationship to the patient (e.g. parent, sibling, friend, …). Toy datasets are available as part of the software package. Missing values are allowed for any of these, but of course not preferred. To protect patient privacy, the software can also input de-identified versions of the input data, but applying date shifting processes may reduce the accuracy of the family predictions.
The parallel processes in Figure 1 show the inference process of family relationships. On the top, FPPA applies decision tree logic for inference of parent–child relationship with basic demographic information. At the bottom, RIFTEHR infers family relationships using emergency contacts. The two methods work simultaneously, and outputs are delivered directly to the next step for family construction, cleaning and reconciliation.
2.2 Family pedigree construction
To increase the software’s flexibility, and as mentioned previously, users have the option to include basic demographic data and/or emergency contact information into E-Pedigrees. FPPA automatically predicts parent–child pairs if users choose to input demographic information only. RIFTEHR can also infer family relationships if the user only inputs emergency contact data. When both data sources are included, family data from one data source can build upon family data from the other. Where relationship conflicts occur, a reconciliation process follows. For example, there may be instances when individual ‘A’ is identified as parent of ‘B’ and individual ‘B’ is identified as parent of ‘C’ according to FPPA whereas RIFTEHR infers ‘A’ is parent of ‘C’. In these instances, the user has the option to prioritize which data source is more accurate. The default is currently set to RIFTEHR. Hence, direct or indirect edges may be added or removed in the graph resulting in merging or splitting of pedigrees. The last step is to ensure pedigrees meet biological assumptions; specifically, each individual node in the graph has at most two in-degree edges from opposite sex individuals (mother and father) and no loops in the pedigree structure. This reconciliation and data cleaning step can be repeated if the user wishes to incorporate additional pedigree data beyond the original inputs. The end output is a cleaned and standardized PED file appropriate for many downstream applications.
3 Results
To demonstrate the advantage of combining FPPA and RIFTEHR into a single application (E-Pedigrees) with additional logic built to combine algorithms, we applied all three prediction methods to Marshfield Clinic’s EHR which captures over 2.6 million patients including 20 000 enrolled in Personalized Medicine Research Project (PMRP) (McCarty et al., 2005). PMRP is a biobank where those enrolled were asked to self-report all first-degree relatives. Of the 20 000 participants, nearly 12 000 self-reported family members that could be mapped to the EHR totaling 16 400 individuals and 4515 families. It is these self-reported families we assumed represented something close to ground truth. Among the over 2.6 million patients, we defined 74 337 nuclear and 12 219 larger than 2-generation families.
We predicted 2667 and 2334 families in PMRP using FPPA and RIFTEHR, respectively. E-Pedigrees showed an improved result with 2787 predicted families (Table 1). We then compared the predicted families with ground truth families assuming (i) family relationships from predicted families (every inferred family relationship, e.g. parent child, siblings, …) should be consistent with family relationships in PMRP. Predicted families containing relationships inconsistent with ground truth were treated as false positives. (ii) Predicted families that maintained a sub-structure of family or a combination of families from PMRP ground truth were treated as true positives. Under these two criterions, applying both methods together in E-pedigrees outperformed baseline method FPPA and RIFTEHR with higher precision (Table 1).
Summary of three methods’ family pedigree prediction relates to PMRP cohort patients
| Method | Number of families | Number of patients | Family size (mean) | Standard deviation |
|---|---|---|---|---|
| FPPA | 2667 True positive: 2441 Precision: 90.4% | 7612 | 2.8 | 1.6 |
| RIFTEHR | 2334 True positive: 2199 Precision: 94.2% | 6835 | 2.9 | 1.5 |
| E-pedigrees | 2787 True positive: 2648 Precision: 95.0% | 7900 | 2.8 | 1.8 |
| Method | Number of families | Number of patients | Family size (mean) | Standard deviation |
|---|---|---|---|---|
| FPPA | 2667 True positive: 2441 Precision: 90.4% | 7612 | 2.8 | 1.6 |
| RIFTEHR | 2334 True positive: 2199 Precision: 94.2% | 6835 | 2.9 | 1.5 |
| E-pedigrees | 2787 True positive: 2648 Precision: 95.0% | 7900 | 2.8 | 1.8 |
Summary of three methods’ family pedigree prediction relates to PMRP cohort patients
| Method | Number of families | Number of patients | Family size (mean) | Standard deviation |
|---|---|---|---|---|
| FPPA | 2667 True positive: 2441 Precision: 90.4% | 7612 | 2.8 | 1.6 |
| RIFTEHR | 2334 True positive: 2199 Precision: 94.2% | 6835 | 2.9 | 1.5 |
| E-pedigrees | 2787 True positive: 2648 Precision: 95.0% | 7900 | 2.8 | 1.8 |
| Method | Number of families | Number of patients | Family size (mean) | Standard deviation |
|---|---|---|---|---|
| FPPA | 2667 True positive: 2441 Precision: 90.4% | 7612 | 2.8 | 1.6 |
| RIFTEHR | 2334 True positive: 2199 Precision: 94.2% | 6835 | 2.9 | 1.5 |
| E-pedigrees | 2787 True positive: 2648 Precision: 95.0% | 7900 | 2.8 | 1.8 |
4 Using the application
Users should follow the instructions to provide basic demographic information and/or emergency contact data. When available, users can also incorporate their own PED files generated from tertiary sources. The python version application is available online (https://github.com/xiayuan-huang/E-pedigrees) for users to download. A detailed user guide is available on the same application download page as well. Users will find E-Pedigrees fast, easy, and flexible with different data sources.
5 Conclusions
We present the publicly available implementation of our automatic family pedigree perdition algorithm. Although we demonstrate E-pedigrees can efficiently and effectively identify family pedigrees with EHR data with improvements from existing methods, limitations remain. For example, identifying adopted relationships will be a challenge. If only basic demographic data is available to the user, E-pedigrees cannot identify parent–child relationships where both individuals do not share an exact last name. Such instances include name hyphenations and name changes due to marriage. E-pedigrees is also sensitive to the longitudinal nature of the input data. This is evident by a propensity for small nuclear families being predicted over three- or more generation families. It is expected that pedigree predictions will improve overtime as EHR systems become more complete and interoperable while users incorporate tertiary data sources into the E-Pedigrees pipeline. Even with these limitations, EHR-derived pedigrees can still be applied by data scientists to study a wide range of human diseases. Given E-Pedigrees’s high accuracy and completeness, such EHR-derived family data could conceivably lead to future prediction tools that capture family histories in real time for clinical care.
Funding
This work was supported in part by National Institute of General Medical Sciences [1R01GM114128 and 1R01GM130715] and National Human Genome Research Institute [1U01HG006389].
Conflict of Interest: none declared.


{kind=link}